JAZMP uvodoma pojasnjuje, da kot pristojni regulatorni in nadzorni organ ne more svetovati zavezancem glede ravnanja v konkretnih primerih oziroma v posameznih postopkih. Odgovori, objavljeni po izobraževanjih, so zato namenjeni splošnemu pojasnjevanju zakonodajnih zahtev in ne predstavljajo pravno zavezujoče razlage predpisov ali vnaprejšnje odločitve JAZMP v morebitnih upravnih ali nadzornih postopkih.
Zavezujočo razlago prava Evropske unije, vključno z Uredbo (EU) 2017/745 o medicinskih pripomočkih (MDR) in Uredbo (EU) 2017/746 o in vitro diagnostičnih medicinskih pripomočkih (IVDR), lahko poda le Sodišče Evropske unije. Avtentično razlago zakonov Republike Slovenije lahko poda Državni zbor Republike Slovenije, neobvezna pojasnila pa lahko poda tudi predlagatelj zakona, tj. Ministrstvo za zdravje.
Glede na navedeno so odgovori JAZMP informativne narave in predstavljajo nezavezujoča strokovna pojasnila, podana na podlagi razpoložljivih informacij ter veljavne zakonodaje v času njihove priprave.
Zaradi boljše berljivosti se izraz »uredbi«, kadar je uporabljen v tem dokumentu, nanaša na Uredbo (EU) 2017/745 o medicinskih pripomočkih (MDR) in Uredbo (EU) 2017/746 o in vitro diagnostičnih medicinskih pripomočkih (IVDR). Kadar so navedeni le členi MDR, se šteje, da so zajeti tudi ustrezni členi IVDR.
A. Splošna vprašanja
1. Ali se beseda »zdravljenje« lahko navaja v označevanju medicinskega pripomočka in v promocijskem gradivu, kadar je to smiselno, čeprav lahko povzroča pomislek, da gre za zdravilo?
Izraz »zdravljenje« se lahko uporabi, kadar je skladen s predvidenim namenom, ki ga je določil proizvajalec, in kadar navedba uporabnika ne zavaja glede narave, lastnosti ali načina delovanja izdelka. MDR že v 2. členu v definiciji medicinskega pripomočka med medicinske namene vključuje tudi zdravljenje oziroma lajšanje bolezni. Ključno je, da pripomoček svojega glavnega predvidenega učinka na človeško telo ne dosega na farmakološki, imunološki ali metabolični način, čeprav so mu ti načini lahko v pomoč pri delovanju. Navedbe v označevanju, navodilih in promocijskem gradivu morajo biti skladne s 7. členom MDR in VIII poglavjem ZMedPri-1 ter dokazanim predvidenim namenom in dokumentacijo proizvajalca
2. Kdaj bo vzpostavljen informacijski sistem JAZMP?
Vzpostavitev informacijskega sistema JAZMP je vezana tudi na razvoj in obvezno uporabo EUDAMED ter nacionalne izvedbene rešitve. Do polne vzpostavitve informacijskega sistema je treba upoštevati navodila JAZMP glede načina posredovanja podatkov in sprememb.
B. Navodila za uporabo
1. Nekateri proizvajalci trdijo, da seznam sestavin lahko ostane v angleškem jeziku, čeprav je medicinski pripomoček namenjen končnemu uporabniku. Ali so pri tem izjeme?
Ne. V skladu s 7. členom ZMedPri-1 morajo biti pri medicinskih pripomočkih, namenjenih končnemu uporabniku oziroma nestrokovnjaku, oznake in navodila za uporabo iz oddelka 23 Priloge I MDR v slovenskem jeziku. Če je seznam sestavin naveden na oznaki, embalaži, v navodilu za uporabo ali drugih informacijah, ki spremljajo pripomoček, mora biti zagotovljen v slovenskem jeziku.
Izjema iz 7. člena ZMedPri-1 se nanaša na pripomočke za profesionalno uporabo in ne na pripomočke, namenjene nestrokovnjakom. Prevod mora vsebinsko ustrezati informacijam proizvajalca.
2. Navodila za uporabo so lahko dolga več deset strani in jih je nemogoče prevesti v 10 dneh. Kaj v tem primeru?
Subjekt mora zagotovitev navodil v slovenskem jeziku načrtovati pravočasno. Pri medicinskih pripomočkih, ki jih je proizvajalec opredelil za profesionalno uporabo, so lahko oznake in navodila za uporabo v slovenskem ali drugem uporabniku razumljivem jeziku.
Če zdravstvena ustanova zahteva navodilo za uporabo v slovenskem jeziku, ga mora subjekt zagotoviti takoj oziroma najpozneje v desetih dneh. Obsežnost navodil subjekta ne razbremeni te obveznosti.
3. Kaj pa v primeru, ko navodil za uporabo ni?
Najprej je treba preveriti, ali so navodila za uporabo za konkreten medicinski pripomoček v skladu z MDR zahtevana.
Navodila za uporabo praviloma priloži proizvajalec. MDR pa v Prilogi I, poglavje III, točka 23.1(d), določa izjemo za pripomočke razreda I in IIa: navodila za uporabo za te pripomočke niso obvezna, če se lahko pripomoček uporablja varno brez takih navodil in če v zahtevah glede informacij, ki jih zagotovi proizvajalec, ni določeno drugače.
Če so navodila za uporabo potrebna za varno in pravilno uporabo pripomočka, jih mora zagotoviti proizvajalec. Uvoznik in distributer morata v okviru svojih obveznosti preveriti, ali so zahtevane informacije priložene pripomočku oziroma dostopne uporabniku.
Če so navodila za uporabo pomanjkljiva ali neustrezna oziroma druge bistvene informacije, ki zagotavljajo varno in pravilno uporabo pripomočka manjkajo, uvoznik ali distributer takega pripomočka ne sme dati na trg oziroma mu omogočiti dostopnosti na trgu, dokler neskladnost ni ustrezno odpravljena.
C. Distributerji, uvozniki in registracija pri JAZMP
1. Kako boste nadzirali distributerje iz drugih držav EU in Amazona, ki ne bodo prijavljeni v vašo bazo, bodo pa prodajali na slovenskem trgu?
Zakonodaja v Republiki Sloveniji navaja, da se v registre poslovnih subjektov priglasijo subjekti s sedežem poslovanja v RS. Ti subjekti so pod nadzorom JAZMP. Nadzor nad subjekti izven RS prevzema država članica EU, v kateri ima poslovni subjekt sedež poslovanja. Če JAZMP zazna neskladnost pripomočka, ki ga na slovenskem trgu omogoča subjekt iz druge države članice, JAZMP v okviru postopkov in ukrepov nadzora trga obvesti tako pristojni organ te države kot druge pristojne organe, ki sodelujejo pri nadzoru skupnega notranjega trga, kjer velja načelo prostega pretoka blaga.
2. Kdo nadzira skladnost z MDR za tuje spletne trgovine izven EU, ki medicinske pripomočke dostavljajo tudi v Slovenijo?
Pri spletnih trgovinah iz tretjih držav je treba presoditi konkretno dobavno verigo in vlogo subjektov v EU. Medicinski pripomočki, ki so dostopni na trgu EU, morajo izpolnjevati zahteve MDR oziroma IVDR, ne glede na način prodaje. JAZMP lahko ukrepa v okviru svojih pristojnosti, zlasti ob zaznavi neskladnosti, prijavi ali v sodelovanju z drugimi pristojnimi organi, na primer carinskimi organi.
3. Če je distributer iz Hrvaške in želi vstopiti na slovenski trg, na primer v veletrgovinah s pripomočki razreda I, kakšne so njegove obveznosti?
Distributer s sedežem v drugi državi članici EU mora izpolnjevati obveznosti distributerja po 14. členu MDR oziroma IVDR. Registracija pri JAZMP je zahtevana za distributerje s sedežem v Republiki Sloveniji, zato se tuji distributer pri JAZMP ne registrira kot slovenski distributer.
4. Kako je v primeru distributerja ali uvoznika iz druge EU države, ki prodaja v Slovenijo? Kako se registrira na JAZMP?
Če ima distributer ali uvoznik sedež v drugi državi članici EU in nima sedeža v Republiki Sloveniji, se pri JAZMP ne registrira kot subjekt s sedežem v Republiki Sloveniji. Mora pa izpolnjevati obveznosti, ki za njegovo vlogo izhajajo iz MDR oziroma IVDR. Distributer mora izpolnjevati obveznosti 14. člena, uvoznik pa obveznosti 13. člena MDR oziroma IVDR, vključno z registracijo gospodarskega subjekta v EUDAMED in navedbo svojega subjekta na embalažo pripomočka, skladno s 13(3) členom MDR in IVDR.
5. Na trg ponujamo medicinske pripomočke proizvajalca iz ZDA. Ali na JAZMP poročamo v tabeli za uvoznike in v tabeli za distributerje? Včasih uvažamo neposredno, včasih pa kupimo od EU posrednika. Kako postopati?
Vloga subjekta se presoja glede na konkretni pripomoček in konkretno dobavno pot. Če subjekt s sedežem v EU pripomoček proizvajalca iz tretje države prvič daje na trg EU, prevzema odgovornosti uvoznika. Če subjekt pripomoček kupi od drugega gospodarskega subjekta v EU, pri čemer je bil pripomoček že dan na trg EU, prevzema obveznosti distributerja.
Isti poslovni subjekt je zato lahko za nekatere pripomočke uvoznik, za druge pa distributer. Obveznosti, prijave in podatke v ustreznih tabelah je treba voditi glede na dejansko vlogo subjekta pri posameznem pripomočku oziroma dobavni poti.
6. Ko uvoznik prvič da na trg nov medicinski pripomoček, komu poroča – JAZMP ali EUDAMED?
Uvoznik mora izpolnjevati obveznosti iz 13. člena MDR oziroma IVDR ter obveznosti registracije v EUDAMED. Od 28. maja 2026 je uporaba modula za registracijo gospodarskih subjektov in modula UDI/registracija pripomočkov v EUDAMED obvezna. Do 28. 5. 2026 se uvoznik s sedežem v Republiki Sloveniji hkrati registrira pri JAZMP kot uvoznik in JAZMP ob vsaki prvi dobavi pripomočka na trg predloži dokumentacijo, s katero izkazuje skladnost pripomočka. Od 28. 5. 2026 registracija na JAZMP in sporočanje podatkov o uvozu ni več potrebna. Mora pa uvoznik urediti povezavo s proizvajalcem, katerega pripomočke daje na trg EU, v Eudamed.
7. Distributer mora enkrat letno potrditi točnost podatkov v informacijskem sistemu JAZMP. Ali nas bo JAZMP pisno pozval, da potrdimo točnost podatkov oziroma spremembe?
Da. Distributer enkrat letno potrdi točnost podatkov na podlagi zahteve v informacijskem sistemu JAZMP, v roku, ki je naveden v zahtevi in ne sme biti krajši od 30 dni.
Ne glede na letno potrditev mora distributer spremembe podatkov oziroma prenehanje omogočanja dostopnosti pripomočka na trgu Republike Slovenije sporočiti najpozneje v osmih dneh od nastanka spremembe.
8. Medicinskih pripomočkov ne bomo več dobavljali lekarnam. Ali smo kot distributer obvezni obvestiti JAZMP?
Če distributer preneha omogočati dostopnost pripomočka na trgu Republike Slovenije ali se spremenijo podatki, posredovani ob registraciji, mora spremembo sporočiti JAZMP najpozneje v osmih dneh od nastanka spremembe.
Če gre le za spremembo posameznega kupca oziroma prodajnega kanala, pri čemer distributer pripomočku še naprej omogoča dostopnost na trgu Republike Slovenije in se podatki, sporočeni ob registraciji subjekta niso spremenili, obveščanje JAZMP ni potrebno.
9. Ali moramo imeti z vsemi kupci medicinskih pripomočkov, na primer zobozdravniki in zobotehniki, sklenjene pogodbe o prodaji medicinskih pripomočkov?
Sklenjena posebna pogodba sama po sebi ni pogoj za prodajo oziroma distribucijo medicinskih pripomočkov. Ne glede na pogodbeno obliko mora subjekt izpolnjevati obveznosti po MDR/IVDR in ZMedPri-1, zlasti glede preverjanja skladnosti, sledljivosti, pogojev skladiščenja in prevoza, pritožb, zapletov in sodelovanja s pristojnimi organi.
D. EUDAMED, UDI, CE in Izjava o skladnosti
1. Kaj naj stori uvoznik, če proizvajalci še niso vnesli medicinskih pripomočkov v EUDAMED in jih tudi še ne bodo do 28. 5. 2026?
Uvoznik mora v skladu s 13. členom MDR/IVDR preveriti, ali so izpolnjene vse obveznosti, ki jih omenjen člen navaja, kot tudi informacije glede registracije gospodarskega subjekta in pripomočka v EUDAMED. Od 28. 5. 2026 je uporaba tudi modula UDI/registracija pripomočkov v EUDAMED obvezna.
Če se pripomoček z določenim UDI-DI prvič daje na trg EU 28. 5. 2026 ali pozneje, mora biti skladno s 29. členom MDR oziroma 26. členom IVDR pred tem vpisan v EUDAMED. Če je bil pripomoček z določenim UDI-DI dan na trg EU že pred 28. 5. 2026 in se bo dajal na trg tudi po tem datumu, mora biti vpisan v EUDAMED najpozneje do 28. 11. 2026.
Zahteva velja le za pripomočke skladne z MDR/IVDR. Pripomočki skladni z eno izmed direktiv v Eudamed ne bodo vpisani, razen v primeru resnega tveganja za uporabnika. Ti pripomočki so lahko zakonito dani na trg v primeru vstopa proizvajalca v certifikacijski postopek pri priglašenem organu za certifikacijo v skladu z eno izmed uredb, kar proizvajalec izkaže s t.i. potrditvenim pismom priglašenega organa, vse do 28. 12. 2028, vezano na razred tveganja pripomočka.
Ne glede na navedeno uvoznik ne sme dati na trg pripomočka, za katerega ve ali utemeljeno domneva, da ni skladen z MDR oziroma IVDR.
2. Ali je kje objavljeno, kako iskati po EUDAMED bazi, certifikatih, UDI in podobno?
Evropska komisija objavlja navodila, gradiva in posnetke izobraževanj za uporabo posameznih modulov EUDAMED. Uporabniki naj spremljajo tudi spletno stran JAZMP, kjer so objavljene povezave in pojasnila glede EUDAMED.
Koristne povezave:
EUDAMED – Actor Module Workshop – Streaming Service of the European Commission
EUDAMED – Devices Module Workshop – Streaming Service of the European Commission
EUDAMED – Certificates Module Workshop – Streaming Service of the European Commission
3. Ali bo za uvoznike po začetku delovanja EUDAMED še potrebno izpolnjevati zahteve iz 3. odstavka 41. člena ZMedPri-1?
41. člen ZMedPri-1 ureja registracijo in obveznosti uvoznikov v prehodnem obdobju do začetka objave v UL Unije, da je EUDAMED polno funkcionalen in poteku roka določenega s Sklep Komisije (EU) 2025/2371.
Do 28. 5. 2026 uvoznik s sedežem v Republiki Sloveniji ob vsaki prvi dobavi pripomočka na trg JAZMP predloži dokumentacijo, s katero izkazuje skladnost pripomočka, kot sta izjava o skladnosti in certifikat, ter sporoči vsak nakup pripomočka, ki mu bo omogočal dostopnost na trgu Republike Slovenije.
Od 28. 5. 2026 registracija uvoznikov pri JAZMP in sporočanje podatkov o prvi dobavi oziroma uvozu pripomočkov JAZMP nista več potrebna. Obveznosti registracije gospodarskih subjektov in pripomočkov se od tega datuma dalje izvajajo v skladu z MDR oziroma IVDR ter v sistemu EUDAMED.
Uvoznik mora biti registriran v modulu za registracijo gospodarskih subjektov (Actor registration module) ter zagotoviti ustrezno povezavo s proizvajalcem, katerega pripomočke daje na trg Evropske unije.
4. Kako lahko spremenimo informacije v EUDAMED, če prenehamo biti uvoznik za določenega proizvajalca?
Vsako spremembo je potrebno urediti v EUDAMED v ustreznem modulu, v skladu z navodili Evropske komisije. Uvoznik mora zagotoviti, da so podatki v EUDAMED točni in posodobljeni.
5. Kakšen UDI je treba hraniti skladno s 5. odstavkom 41. člena ZMedPri-1 – Basic UDI-DI ali UDI-DI – in koliko časa je treba hraniti to tabelo? Ali bo to potrebno tudi po vzpostavljenem EUDAMED?
Po 5. odstavku 41. člena ZMedPri-1 uvozniki hranijo UDI za pripomočke, ki so jim bili dobavljeni, ne glede na razred tveganja. Ker UDI sestavljata UDI-DI in UDI-PI, je treba hraniti UDI dobavljenega pripomočka, ne le Basic UDI-DI. Basic UDI-DI (Osnovni UDI-DI) je identifikator na višji ravni, ki združuje skupino pripomočkov istega razreda tveganja, predvidenega namena, dizajna in karakteristike izdelave ter je kot tak naveden tako na Izjavi EU o skladnosti kot certifikatu EU priglašenega organa.
ZMedPri-1 za to evidenco ne določa posebnega roka hrambe. Uvoznik mora po 13. členu MDR hraniti izjavo EU o skladnosti in, kadar je ustrezno, certifikat v obdobju iz 10. člena MDR, torej praviloma najmanj 10 let, pri pripomočkih za vsaditev pa najmanj 15 let po tem, ko je bil zadnji pripomoček dan na trg.
6. Ali mora biti UDI naveden na zunanji ovojnini, na primer škatlici, ali je dovolj, da je naveden na stični ovojnini, na primer tubi?
UDI mora biti naveden v skladu s prilogo VI uredb MDR in IVDR. Praviloma mora biti nosilec UDI naveden na označbi ali na pripomočku samem ter na vseh višjih ravneh pakiranja. Odgovornost za dodelitev in namestitev UDI nosi proizvajalec. Distributer ali uvoznik mora pri preverjanju skladnosti upoštevati zahteve za konkretni pripomoček in podatke proizvajalca.
7. Če ima medicinski pripomoček izjavo in certifikat po novi zakonodaji, UDI pa je naveden v dokumentaciji, ne pa na samem pripomočku, ali je tak pripomoček skladen (datuma proizvodnje ne poznamo)?
Zahteva za namestitev nosilca UDI na označbo oziroma pakiranje velja že za večino medicinskih pripomočkov in IVD pripomočkov. Izjemoma so le IVD pripomočki razreda tveganja A ter pripomočki, ki jih je proizvajalec opredelil za večkratno uporabo razreda I, kjer rok za označevanje poteče 26. 5. 2027.
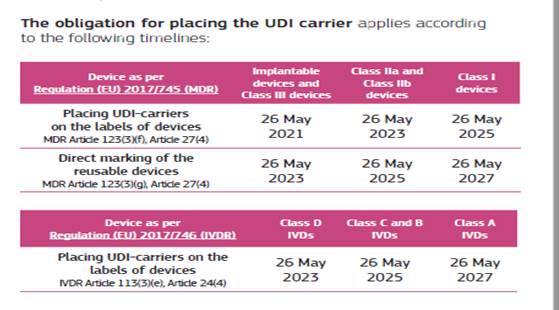
Če UDI manjka tam, kjer bi moral biti naveden, navedba UDI samo v dokumentaciji ne zadošča. Distributer oziroma uvoznik mora pridobiti pojasnilo proizvajalca in pred nadaljnjim omogočanjem dostopnosti preveriti skladnost označevanja.
8. Kako je s sledljivostjo UDI – ali mora biti UDI prisoten tudi v spremnih dokumentih?
MDR in IVDR ne določata splošne zahteve, da mora biti UDI naveden na vseh spremnih dokumentih. UDI mora biti zagotovljen v skladu z zahtevami za sistem UDI iz Priloge VI MDR/IVDR, zlasti glede označevanja oziroma namestitve nosilca UDI in registracije pripomočka, kadar je to ustrezno. Distributerji in uvozniki pa morajo zagotoviti ustrezno sledljivost pripomočkov v dobavni verigi.
Zato morajo razpolagati s podatki, ki omogočajo povezavo dobavljenega pripomočka z gospodarskim subjektom, ki jim je pripomoček neposredno dobavil, ter z gospodarskim subjektom, zdravstveno ustanovo ali zdravstvenim delavcem, ki so mu pripomoček neposredno dobavili, kadar je to zahtevano. Te zahteve postavljata že uredbi, MDR v 25. členu in IVDR v 22. členu – Identifikacija v dobavni verigi. Pri uvoznikih je treba upoštevati tudi obveznost hrambe UDI za pripomočke, ki so jim bili dobavljeni.
9. Kako predlagate, da uvozniki preverjamo in hranimo oziroma dokazujemo preverjanje podatkov proizvajalcev, SRN številk in UDI-DI v EUDAMED, ko gre v praksi za velike količine dokumentov?
Zakonodaja ne predpisuje enotnega obrazca ali oblike evidence. Uvoznik mora vzpostaviti tak sistem, da lahko dokaže izpolnjevanje svojih obveznosti po MDR oziroma IVDR in ZMedPri-1. To je lahko elektronska evidenca, kontrolni seznam, shranjen izpis iz EUDAMED, povezava na zapis v EUDAMED ali drug sledljiv zapis.
Pomembno je, da je razvidno, kaj je bilo preverjeno, kdaj, za kateri pripomoček in kakšen je bil rezultat preverjanja. Pri uvoznikih metoda vzorčenja ne nadomešča obveznosti preverjanja pred dajanjem pripomočka na trg.
10. Ali imajo certifikati in izjave o skladnosti za medicinske pripomočke predpisano obliko?
Izjava EU o skladnosti mora vsebovati podatke, določene v Prilogi IV MDR oziroma IVDR, ni pa predpisana ena sama grafična oblika dokumenta. Tudi certifikati priglašenih organov nimajo enotne postavitve, morajo pa vsebovati vse zahtevane podatke Priloge XII MDR/IVDR.
Pri presoji je zato pomembna predvsem vsebina dokumenta, ne njegova oblika.
11. Ali je navedba standardov v izjavi o skladnosti potrebna, če so standardi navedeni v tehnični mapi pripomočka?
Ne nujno. Priloga IV MDR/IVDR določa obvezno vsebino izjave EU o skladnosti. Med obveznimi elementi je sklic na uporabljene skupne specifikacije, kadar so bile uporabljene, ne pa izrecna obvezna navedba vseh harmoniziranih standardov.
Harmonizirani standardi so praviloma navedeni in utemeljeni v tehnični dokumentaciji proizvajalca. V izjavi EU o skladnosti se lahko navedejo kot dodatna informacija, zlasti kadar se proizvajalec nanje sklicuje pri dokazovanju skladnosti, vendar to ne nadomesti obveznih elementov iz Priloge IV.
Uvoznik oziroma distributer zato preveri predvsem, ali izjava vsebuje vse obvezne podatke iz Priloge IV, vključno s podatki o proizvajalcu, identifikaciji pripomočka, osnovnem UDI-DI, razredu tveganja, navedbi skladnosti z MDR/IVDR, morebitnih skupnih specifikacijah, podatkih o priglašenem organu, kadar je to ustrezno, ter kraju, datumu in podpisu.
12. Kateri certifikati so potrebni za različne razrede medicinskih pripomočkov oziroma IVD pripomočkov? Ali je to glede na razrede kje posebej navedeno?
Vrsta certifikata ni določena samo glede na razred pripomočka, temveč predvsem glede na postopek ugotavljanja skladnosti priglašenega organa, ki je dogovorjen s proizvajalcem ob podpisu sporazuma o certifikaciji. Ti postopki so za medicinske pripomočke določeni v MDR, zlasti v členu 52 in prilogah IX, X in XI, za IVD pripomočke pa v IVDR, zlasti v členu 48 in prilogah IX, X in XI.
Pri medicinskih pripomočkih razreda I certifikat priglašenega organa ni potreben. Tudi pripomočke razreda tveganja I, ki so sterilni ali imajo merilno funkcijo oziroma so s strani proizvajalca namenjeni za večkratno uporabo, spremlja certifikat priglašenega organa. Prav tako je certifikat EU potreben za pripomočke razredov tveganja IIa, IIb in III. Pri IVD pripomočkih certifikat priglašenega organa ni potreben le za razred A. Pripomočki, razvrščeni v razred tveganja A-sterilno spremlja certifikat priglašenega organa, kot tudi pripomočke razredov B, C in D.
Ali bo v konkretnem primeru izdan certifikat za sistem vodenja kakovosti, certifikat za oceno tehnične dokumentacije ali druga vrsta certifikata, je odvisno od izbranega oziroma zahtevanega postopka ugotavljanja skladnosti in obsega presoje priglašenega organa. Zato uvoznik oziroma distributer ne preverja le ali certifikat obstaja, temveč ali predloženi certifikat po obsegu zajema konkretni pripomoček, njegovo razvrstitev, proizvajalca, uporabljeni postopek ugotavljanja skladnosti in veljavno izjavo EU o skladnosti.
13. Kaj vse moramo kot distributer preveriti na izjavi o skladnosti oziroma certifikatu? Ali je dovolj, da preverimo, da dokumentacija obstaja?
Ne. Distributer ne preveri le, ali dokumentacija obstaja, temveč mora v okviru 14. člena MDR oziroma IVDR preveriti, da je pripomoček označen z oznako CE, da je pripravljena izjava EU o skladnosti, da so priložene zahtevane informacije, da je naveden uvoznik, kadar je to relevantno, in da je dodeljen UDI, kjer je to zahtevano.
Pri izjavi EU o skladnosti je treba preveriti, ali se nanaša na pravi pripomoček in proizvajalca ter ali vsebuje obvezne podatke iz Priloge IV. Če je za pripomoček potreben certifikat priglašenega organa, je treba preveriti njegovo veljavnost, obseg, priglašeni organ ter skladnost podatkov z izjavo EU o skladnosti in označevanjem.
Distributer ne presoja celotne tehnične dokumentacije, mora pa zaznati očitna neskladja. Če ugotovi ali utemeljeno sumi neskladnost, pripomočka ne sme omogočiti na trgu, dokler neskladnost ni odpravljena.
V pomoč je na spletni strani JAZMP objavljeno vodilo za pregled skladnosti medicinskega pripomočka.
14. Če je certifikat naveden v izjavi EU o skladnosti ali mora distributer preveriti tudi certifikat ali je to le priporočljiva dobra praksa?
Distributer mora preveriti izpolnjevanje zahtev iz 14. člena MDR/IVDR in ne sme omogočiti dostopnosti pripomočka, če meni ali utemeljeno domneva, da pripomoček ni skladen. MDR/IVDR pri preverjanju izrecno omenjata izjavo EU o skladnosti, vendar je certifikat, kadar je za pripomoček zahtevan in naveden, pomemben del dokazovanja skladnosti. Zato je preverjanje relevantnih podatkov certifikata praviloma del skrbnega preverjanja dokumentacije, zlasti pri pripomočkih, pri katerih sodeluje priglašeni organ ali kadar obstaja dvom o skladnosti. Distributer mora v okviru skrbnega preverjanja preveriti vsaj osnovne podatke certifikata, zlasti njegovo veljavnost, obseg, proizvajalca, pripomoček oziroma skupino pripomočkov in skladnost z izjavo EU o skladnosti.
V pomoč je na spletni strani JAZMP objavljeno vodilo za pregled skladnosti medicinskega pripomočka.
15. Če ima proizvajalec na trgu MDR skladen pripomoček razreda IIa, hkrati pa je na trgu še »legacy device« istega izdelka, kakšna izjava o skladnosti velja za »legacy device«?
Za »legacy device« se uporablja izjava o skladnosti, pripravljena po prejšnji zakonodaji, torej po MDD oziroma AIMDD, če pripomoček izpolnjuje pogoje za prehodno obdobje po MDR. Dejstvo, da ima proizvajalec za isti ali podoben pripomoček tudi MDR skladno različico, ne spremeni dokumentacije, ki velja za »legacy device«.
Pri preverjanju je treba ločiti med MDR skladnim pripomočkom in »legacy device« ter preveriti, katera izjava, certifikat in označevanje se nanašajo na konkretni pripomoček oziroma njegovo različico. Vedno je treba preveriti tudi datum dajanja na trg, veljavnost dokumentacije, pogoje prehodnih določb in ali pri pripomočku ni prišlo do pomembnih sprememb zasnove ali predvidenega namena. Ob tem izpostavljamo tudi, da so pripomočki razreda tveganja I vse od začetka uporabe MDR, od 26. 5. 2021, lahko dani na trg le skladni z MDR, kar posledično pomeni, ustrezno pripravljeno Izjavo EU o skladnosti.
E. Vzorčenje, sledljivost in evidence
1. Kako je z obveznostjo vzorčenja medicinskih pripomočkov pri distributerju oziroma uvozniku? Ali pregled fotografij, ki jih posreduje proizvajalec, zadošča?
Za distributerja MDR/IVDR dopuščata reprezentativno metodo vzorčenja za preverjanja skladnosti zahtev uredb iz 14. člena. Fotografije lahko pomagajo pri predhodnem preverjanju, vendar ne nadomestijo pregleda dobavljenega pripomočka, saj na fotografijah pogosto niso razvidni vsi podatki, kot so lot, rok uporabe, UDI, konkretna označba ali stanje embalaže. Uvoznik mora pred dajanjem pripomočka na trg preveriti zahteve iz 13. člena MDR/IVDR; zanj za preverjanje skladnosti pripomočka metoda vzorčenja ne velja..
2. Kolikšen delež medicinskih pripomočkov se vzorči na letni ravni glede na zalogo?
Predpis ne določa enotnega odstotka, ki bi veljal za vse distributerje. Reprezentativni vzorec mora subjekt določiti na podlagi ocene tveganja, obsega dobav, števila proizvajalcev in dobaviteljev, vrste pripomočkov, razredov tveganja, zgodovine neskladnosti in drugih relevantnih okoliščin. Metoda vzorčenja mora biti opisana v internem postopku in mora omogočati dokazovanje, da so preverjanja iz 14. člena MDR/IVDR dejansko izvedena.
3. Ali je pri vzorčenju treba dejansko odpreti pripomoček ali zadošča preverjanje elektronske dokumentacije?
Obseg pregleda je odvisen od pripomočka, embalaže, tveganja in podatkov, ki jih je treba preveriti. Ni dopustno odpirati sterilne ali varnostno zaprte embalaže, če bi se s tem ogrozila skladnost, sterilnost ali uporabnost pripomočka. Če je mogoče vse zahtevane informacije preveriti na zunanji embalaži, označbi, spremni dokumentaciji in elektronski dokumentaciji, odpiranje ni potrebno. Kadar potrebnih informacij ni mogoče preveriti, mora subjekt od dobavitelja ali proizvajalca pridobiti ustrezna dokazila oziroma drugače zagotoviti skladnost preverjanja.
4. Če proizvajalec nalepi varnostno nalepko proti odpiranju in je distributerju onemogočeno vzorčenje notranje sterilne embalaže ali navodil, kaj mora storiti?
Distributer ne sme odpreti embalaže, če bi s tem posegel v sterilnost, varnostni pečat ali skladnost pripomočka. V takem primeru preveri zunanje oznake, spremno dokumentacijo in dokumentacijo proizvajalca ter od proizvajalca ali dobavitelja pridobi dokazila o notranji vsebini, navodilih ali oznakah, ki jih ni mogoče preveriti brez posega. Postopek naj bo opisan v internem sistemu kakovosti oziroma postopku vzorčenja.
5. Če je pregledan vzorec skladen, ali ga je treba shraniti ali se lahko vrne v prodajo?
Če je bil pripomoček pregledan brez posega, ki bi ogrozil njegovo skladnost, varnost, sterilnost, embalažo ali pogoje proizvajalca, se lahko vrne v prodajo. Subjekt mora hraniti zapis o izvedenem preverjanju. Če je bila embalaža poškodovana, varnostni pečat odprt ali je drugače poseženo v pripomoček, ga ni dopustno vrniti v običajno distribucijsko verigo, razen če proizvajalec potrdi, da je pripomočke še vedno skladen, varen in učinkovit..
6. Na kakšen način moramo uvozniki in distributerji skrbeti za evidence o prodanih medicinskih pripomočkih?
Uvozniki in distributerji morajo voditi evidence na način, ki omogoča ustrezno sledljivost pripomočkov in dokazovanje izpolnjevanja njihovih obveznosti po MDR/IVDR.
V skladu s 25. členom MDR morajo gospodarski subjekti za predpisano obdobje pristojnemu organu omogočiti identifikacijo vsakega gospodarskega subjekta, ki jim je pripomoček neposredno dobavil, vsakega gospodarskega subjekta, ki so mu pripomoček neposredno dobavili, ter vsake zdravstvene ustanove ali zdravstvenega delavca, ki so mu pripomoček neposredno dobavili.
Iz evidenc naj bodo zato razvidni najmanj podatki o pripomočku, dobavitelju, prejemniku, datumu dobave in relevantnih identifikacijskih podatkih pripomočka, kot so oznaka pripomočka, serija ali lot, kadar je relevantno, ter UDI, kadar se uporablja.
Obseg evidence je odvisen od vloge subjekta, vrste pripomočka in zahtev glede sledljivosti. Posebno pozornost je treba nameniti pripomočkom za vsaditev in pripomočkom višjih razredov tveganja, pri katerih je sledljivost ključna za učinkovito izvajanje morebitnih varnostnih korektivnih ukrepov, odpoklicev ali drugih ukrepov za varovanje zdravja pacientov in uporabnikov.
Uvozniki in distributerji morajo poleg tega voditi tudi evidence oziroma registre, ki jih za njihovo vlogo posebej določata 13. in 14. člen MDR/IVDR, na primer register pritožb, neskladnih, odpoklicanih in umaknjenih pripomočkov.
7. Ali je treba voditi serijske številke medicinskih pripomočkov?
Če je serijska številka del identifikacije pripomočka in je pomembna za sledljivost, jo je treba voditi v obsegu, ki omogoča izpolnjevanje obveznosti dobavne verige, zlasti v primeru pritožb, zapletov, odpoklicev ali varnostnih korektivnih ukrepov. Pri pripomočkih, ki imajo UDI-DI določen, že UDI-PI, kot del UDI-DI modela pripomočka, opredeljuje proizvodno enoto pripomočka. Pri pripomočkih za vsaditev in drugih visoko tveganih pripomočkih so zahteve sledljivosti strožje.
8. Proizvajalec je ukinil serijske številke na pripomočkih. Kako je s sledljivostjo?
Če proizvajalec spremeni način identifikacije pripomočka, mora še vedno zagotoviti sledljivost v skladu z MDR/IVDR in sistemom UDI. Distributer ali uvoznik mora preveriti, kateri identifikatorji so na pripomočku uporabljeni, na primer UDI-DI, ki že vključuje tudi proizvodno enoto pripomočka, lot ali serijska številka, ter temu prilagoditi svoje evidence. Če sprememba onemogoča sledljivost ali vzbuja dvom o skladnosti, je treba od proizvajalca pridobiti pojasnilo in po potrebi zadržati nadaljnjo dobavo.
9. Ali je dovolj, da imamo v tabeli pripomočke, ki so v naši ustanovi vodeni kot medicinski pripomočki in podatke, kaj moramo preveriti? Ali lahko uvoznik izpolni to tabelo ob prijavi na razpis?
Tabela ali elektronska evidenca je lahko ustrezna, če vsebuje dovolj podatkov za dokazovanje izvedenih preverjanj in sledljivosti. Priporočljivo je, da vsebuje vsaj naziv pripomočka, proizvajalca, uvoznika, razred tveganja, identifikatorje, relevantno dokumentacijo, status preverjanja, datum preverjanja in odgovorno osebo. Vprašanja glede prilog k razpisom in zahtev posameznih naročnikov niso predmet MDR/IVDR, temveč pravil javnega naročanja oziroma razpisne dokumentacije.
F. Prevodi, prepakiranje, pritožbe in vigilanca
1. Kakšni so kriteriji za »prevajalca« pri distributerju po 16. členu MDR/IVDR? Ali je to lahko izkušena oseba z naravoslovno izobrazbo, ki prevaja tudi informacije/navodila za zdravila?
MDR/IVDR ne določata posebne formalne izobrazbe ali naziva osebe, ki izvaja prevajanje informacij o medicinskem pripomočku.
Kadar distributer ali uvoznik zagotavlja prevod informacij, ki jih je pripravil proizvajalec, za namen omogočanja dostopnosti pripomočka v Republiki Sloveniji, mora zagotoviti, da je prevod natančen, popoln, sledljiv in redno posodobljen ter za navedeno pridobiti certifikat priglašenega organa za vzpostavljen sistem vodenja kakovosti, v skladu s 4. odstavkom 16. člena MDR Prevod ne sme spreminjati predvidenega namena pripomočka ali vsebine informacij proizvajalca.
Če dejavnost spada v okvir 16. člena MDR/IVDR, mora distributer oziroma uvoznik izpolniti tudi zahteve iz tretjega in četrtega odstavka 16. člena MDR/IVDR, vključno z obvestilom proizvajalcu in pristojnemu organu najmanj 28 dni pred omogočanjem dostopnosti pripomočka ter predložitvijo že omenjenega certifikata priglašenega organa pristojnemu organu. Dodatna pojasnila glede izvajanja dejavnosti prepakiranja in ponovnega označevanja po 16. členu MDR/IVDR so podana v smernici MDCG 2021-26.
2. Kako se zagotovi sistem kakovosti v zvezi s prepakiranjem?
Distributer ali uvoznik, ki izvaja prepakiranje oziroma ponovno označevanje v obsegu 16. člena MDR/IVDR, mora imeti vzpostavljen sistem vodenja kakovosti v skladu s tretjim odstavkom 16. člena MDR/IVDR.
Sistem mora zagotavljati, da se dejavnosti izvajajo pod pogoji, ki ohranjajo prvotno stanje pripomočka, ter da embalaža prepakiranega pripomočka nima napak, ni slabe kakovosti ali neurejena. Pri sterilnih pripomočkih je treba posebej upoštevati, da se prvotno stanje ne ohrani, če je embalaža, potrebna za ohranjanje sterilnosti, odprta, poškodovana ali drugače prizadeta.
Distributer ali uvoznik mora najmanj 28 dni pred omogočanjem dostopnosti na novo označenega ali prepakiranega pripomočka obvestiti proizvajalca in pristojni organ ter pristojnemu organu predložiti certifikat priglašenega organa o skladnosti sistema vodenja kakovosti z zahtevami 16. člena MDR/IVDR. Dodatna pojasnila glede izvajanja teh dejavnosti so podana v smernici MDCG 2021-26.
3. Kje postaviti mejo med reklamacijo in zapletom? Ali je vsaka reklamacija tudi zaplet, na primer poškodba med transportom?
Reklamacije oziroma pritožbe so širši pojem. Lahko so kakovostne, lahko so povezane z neskladnostjo, lahko pa so tudi varnostne in pomenijo poročilo oziroma sum na zaplet.
Vsaka reklamacija zato ni nujno zaplet. Reklamacija se lahko nanaša na pomanjkljivo kakovost, poškodovano embalažo, transportno poškodbo, napako pri dobavi, označevanju, dokumentaciji ali delovanju pripomočka. Vendar lahko pomanjkljiva kakovost v določenih okoliščinah povzroči ali bi lahko povzročila zaplet. Zato je treba vsako pritožbo oceniti tudi z vidika morebitnega vpliva na varnost pacienta, uporabnika ali druge osebe.
Primer transportne poškodbe sam po sebi še ne pomeni nujno zapleta. Če je poškodba ugotovljena pred uporabo, pripomoček ni bil uporabljen in ni vpliva na varnost ali učinkovitost, gre praviloma za kakovostno reklamacijo oziroma neskladnost. Če pa transportna poškodba lahko vpliva na sterilnost, delovanje, varnost ali pravilno uporabo pripomočka, jo je treba obravnavati tudi z vidika suma na zaplet oziroma resnega tveganja.
V skladu s petim odstavkom 14. člena MDR/IVDR morajo distributerji, ki prejmejo pritožbe ali poročila zdravstvenih delavcev, pacientov ali uporabnikov o sumih na zaplete v zvezi s pripomočkom, katerega dostopnost so omogočili, te informacije takoj poslati proizvajalcu ter po potrebi pooblaščenemu predstavniku proizvajalca in uvozniku. Voditi morajo register pritožb, neskladnih, odpoklicanih in umaknjenih pripomočkov ter proizvajalca in po potrebi pooblaščenega predstavnika in uvoznika obveščati o takšnem spremljanju ter jim na zahtevo sporočati vse informacije.
4. Pri poročanju zapletov proizvajalcem prihaja do podvajanja poročanja v distribucijski verigi. Ali morajo vsi distributerji poročati neposredno proizvajalcu?
MDR/IVDR določata, da distributerji, ki prejmejo pritožbe ali poročila o sumih na zaplete, te informacije takoj pošljejo proizvajalcu ter po potrebi pooblaščenemu predstavniku in uvozniku. JAZMP ne ureja pogodbenih razmerij med distributerji. Subjekti lahko v pogodbah in postopkih določijo praktične poti obveščanja, vendar mora biti zagotovljeno, da proizvajalec informacijo prejme pravočasno in da zaradi interne ureditve ne pride do zamude pri obravnavi zapleta ali varnostnega tveganja.
5. Kako mora pooblaščeni servis ravnati, ko prejme medicinski pripomoček z očitnimi znaki nepooblaščenega posega, na primer z vgrajenimi ponarejenimi rezervnimi deli?
Če je servis hkrati distributer ali drug gospodarski subjekt po MDR/IVDR, mora ravnati v skladu s svojimi obveznostmi glede neskladnih ali domnevno ponarejenih pripomočkov.
O ugotovitvah obvestite proizvajalca oziroma pooblaščenega predstavnika ali distributerja, zadržite nadaljnjo uporabo ali vračilo v promet, kadar obstaja tveganje in voditi zapis o ugotovitvah. Če obstaja sum na ponarejen pripomoček ali resno tveganje, se o tem obvesti tudi JAZMP.
Zaznano neskladnost ali kršitev lahko prijavite JAZMP preko spletnega obrazca ali na elektronski naslov .
Pravne ali fizične osebe, ki menijo, da medicinski pripomoček pomeni resno tveganje ali da je ponarejen poročajo JAZMP preko obrazca na elektronski naslov .
G. Lekarne, naročilnice in zdravstvene ustanove
1. Če distributer že preverja skladnost in če pripomoček ne ustreza, ga ne sme prodati naprej, zakaj morajo lekarne ponovno preverjati?
MDR in IVDR obveznosti nalagata vsakemu gospodarskemu subjektu v dobavni verigi. Tudi lekarna, kadar nastopa kot distributer, mora pri omogočanju dostopnosti pripomočka ravnati s potrebno skrbnostjo in izvajati obveznosti distributerja. Preverjanje se lahko organizira sorazmerno tveganju, z uporabo reprezentativne metode vzorčenja, vendar odgovornost posameznega distributerja v verigi ni v celoti prenesena na predhodnega distributerja.
2. Kako je z izdajo oblog za rane, obližev in podobnih pripomočkov po kosih v lekarnah, ne kot celih škatel?
Izdaja oziroma prodaja po kosih je dopustna, kadar je skladna s predvidenim namenom, navodili proizvajalca, označevanjem in varnostjo pripomočka. Če proizvajalec predvidi pakiranje na način, iz katerega je razvidno, da so posamezni kosi namenjeni samostojni izdaji, na primer 50 x 1 kos, je takšna izdaja lahko skladna, če so uporabniku zagotovljene vse potrebne informacije za varno uporabo in je zagotovljena sledljivost pripomočka.
Če pri izdaji po kosih ne pride do posega v zunanjo embalažo pripomočka, ki jo je proizvajalec predvidel za končnega uporabnika, oziroma do prepakiranja ali ponovnega označevanja, se takšno ravnanje praviloma ne šteje za prepakiranje ali ponovno označevanje po 16. členu MDR.
Če pa bi bila za izdajo po kosih potrebna sprememba zunanje embalaže, sprememba velikosti pakiranja, prepakiranje, ponovno označevanje, dodajanje ali prevajanje informacij, mora lekarna oziroma distributer presoditi uporabo 16. člena MDR in izpolniti ustrezne zahteve.
3. Kaj če proizvajalec ne more izdati izjave, da se pripomoček lahko prodaja po kosih? Ali je prodaja vseeno dovoljena?
Če proizvajalec prodaje po kosih ni predvidel in ne more potrditi, da je tak način prodaje skladen, lekarna oziroma distributer ne sme samovoljno spremeniti načina omogočanja dostopnosti pripomočka na trgu.
V takem primeru je prodaja posameznih kosov lahko dopustna le, če je iz pakiranja, označevanja, navodil ali drugih informacij proizvajalca razvidno, da so posamezne enote namenjene samostojni izdaji oziroma uporabi. Če to ni razvidno, je treba pripomoček izdajati v skladu z navodili proizvajalca in zahtevami za konkretni pripomoček oziroma pridobiti dodatno pojasnilo proizvajalca ali dobavitelja.
4. Katere dokumente oziroma informacije mora imeti lekarna pri izdaji medicinskih pripomočkov po kosih?
Ali mora lekarna pridobiti izjavo proizvajalca o možnosti izdaje po kosih? Ali je dovolj kopija slovenskega dela navodil za uporabo? Ali se lahko navodila prepišejo v drug dokument, npr. Word, če se vsebina ne spremeni?
MDR ne predpisuje enotnega obrazca za izjavo proizvajalca o možnosti izdaje medicinskega pripomočka po kosih. Če se taka izjava pridobi, naj bo iz nje jasno razvidno, na kateri pripomoček oziroma pakiranje se nanaša, ali je izdaja po kosih dopustna in pod kakšnimi pogoji ter kakšen način dajanja na trg je predvidel proizvajalec, predvsem glede navodil za uporabo in skladnosti s Prilogo I MDR/IVDR – Splošne zahteve glede varnosti in učinkovitosti.
Lekarna mora uporabniku zagotoviti vse informacije, potrebne za varno uporabo pripomočka. Kopija slovenskega dela navodil za uporabo lahko zadošča, če vsebuje vse relevantne informacije proizvajalca in je popolna, pravilna ter čitljiva. Če so za varno uporabo pomembni tudi simboli, slike, opozorila ali drugi deli navodil, morajo biti zagotovljeni tudi ti.
Prepis navodil v drug dokument, npr. Word, je lahko sprejemljiv, če se spremeni samo oblika, ne pa vsebina, in če se pri tem ne izgubijo ali spremenijo pomembne informacije, simboli ali opozorila. Z navedenim mora biti seznanjen tudi proizvajalec in se s predlogom strinjati.
Če izdaja po kosih vključuje spremembo zunanje embalaže, prepakiranje, ponovno označevanje, dodajanje ali prevajanje informacij, je treba upoštevati tudi zahteve iz 16. člena MDR.
5. Kako je z deljenjem vzorcev medicinskih pripomočkov pacientom oziroma strankam v lekarni in zakaj so potrebne posebne evidence?
Deljenje vzorcev medicinskih pripomočkov splošni javnosti ureja prvi odstavek 24. člena ZMedPri-1. Vzorec medicinskega pripomočka mora izpolnjevati zahteve, ki veljajo za pripomoček, biti označen z navedbo, da gre za vzorec, in biti v najmanjšem pakiranju.
Pri deljenju vzorcev pacientom oziroma strankam v lekarni mora poslovni subjekt voditi evidenco o vrsti in količini razdeljenih vzorcev. Namen evidence je zagotavljanje sledljivosti, saj se tudi pri vzorcih lahko pojavijo pritožbe, zapleti, odpoklici ali varnostni korektivni ukrepi.
6. Na naročilnice ZZZS se izdajajo pripomočki po kosih, na primer igle, brizge, gaze, katetri, vrečke za seč. Od vseh zastopnikov oziroma proizvajalcev ne dobimo izjav o dovoljenem prepakiranju. Kako ravnati?
Vprašanje predpisovanja oziroma izdaje na naročilnice ZZZS ni v pristojnosti JAZMP. Z vidika zakonodaje o medicinskih pripomočkih pa mora biti vsak pripomoček, ki se izda uporabniku, skladen, sledljiv in opremljen z informacijami, potrebnimi za varno uporabo. Če se pripomoček vzame iz večjega pakiranja, mora biti tak način izdaje skladen z navodili proizvajalca ali z izrecnim dovoljenjem proizvajalca ter ne sme ogroziti varnosti, sterilnosti ali sledljivosti.
7. Kaj vse mora imeti lekarna glede medicinskih pripomočkov shranjeno oziroma dokumentirano za potrebe inšpekcijskega nadzora?
Lekarna mora imeti dokumentacijo in evidence, s katerimi lahko izkaže izpolnjevanje obveznosti distributerja iz 14. člena MDR/IVDR: postopke preverjanja skladnosti, dokazila o izvedenih preverjanjih oziroma vzorčenju, sledljivost, pogoje skladiščenja in prevoza, obravnavo pritožb in zapletov, evidence neskladnih, umaknjenih in odpoklicanih pripomočkov ter dokazila o osebah, odgovornih za skladnost, vigilanco in strokovno svetovanje, kadar so zahtevane, kot tudi preverjanja ali je uvoznik v zvezi z uvoženimi pripomočki izpolnil zahteve iz člena 13(3).
8. Ali morajo biti lekarne registrirane v EUDAMED?
Lekarna se kot distributerji medicinskih pripomočkov ne registrirajo v EUDAMED. Lekarna s sedežem v Republiki Sloveniji, ki omogoča dostopnost medicinskih pripomočkov, pa se mora kot distributer registrirati pri JAZMP.
9. Zakaj morajo lekarne voditi svoje evidence certifikatov in dokumentacije, če proizvajalec že zagotavlja skladnost izdelka ob prihodu na trg?
Obveznosti so določene za celotno dobavno verigo, saj vsak gospodarski subjekt odgovarja za svojo vlogo pri omogočanju dostopnosti medicinskih pripomočkov na trgu. Namen teh obveznosti je preprečiti, da bi neskladni pripomočki prišli do uporabnikov, ter zagotoviti sledljivost, učinkovito ukrepanje ob odpoklicih in obravnavo zapletov.
Lekarna kot distributer mora pred omogočanjem dostopnosti pripomočka preveriti, da so izpolnjene zahteve, ki veljajo za distributerje. Obseg preverjanja se lahko, kadar je to dopustno, prilagodi z uporabo reprezentativne metode vzorčenja, vendar mora lekarna imeti dokazila, da je svoje obveznosti kot distributer izpolnila.
10. Kako dolgo se morajo v lekarni hraniti certifikati in izjave o skladnosti?
Za distributerje MDR/IVDR ne določata enake splošne obveznosti hrambe izjav in certifikatov kot za uvoznike. Distributer mora imeti na voljo razpoložljive informacije in dokumentacijo, s katerimi lahko na zahtevo pristojnega organa dokaže skladnost pripomočka oziroma izpolnjevanje svojih obveznosti. Dokumentacija je lahko hranjena tudi elektronsko ali dostopna preko dobavitelja, če je dostop zanesljiv in pravočasen.
11. Ali morajo lekarne shranjevati izjave in certifikate ali je dovolj, da imajo dostop do njih?
Dokumentacija je lahko v digitalni obliki oziroma dostopna preko dobavitelja ali drugega zanesljivega sistema, če lekarna lahko ob nadzoru ali obravnavi neskladnosti dokumentacijo pridobi pravočasno. Ni nujno, da se dokumentacija hrani v papirni obliki na vsaki enoti, razen če to izhaja iz internih postopkov ali posebnih zahtev. Pomembno je, da je odgovornost in dostopnost dokumentacije jasno urejena.
12. Če je treba shranjevati DoC in EC certifikate kot lekarna oziroma specializirana prodajalna, ali morajo biti shranjeni na vseh enotah ali je dovolj na sedežu?
Zakonodaja ne zahteva nujno hrambe papirnih kopij na vsaki posamezni enoti. Dokumentacija je lahko organizirana centralno, če imajo enote zagotovljen pravočasen dostop do relevantnih dokumentov in če je ob nadzoru mogoče izkazati skladnost ter sledljivost za pripomočke, ki jih enota omogoča uporabnikom. Interni postopki morajo jasno določati, kje se dokumentacija hrani in kdo je odgovoren za dostop.
13. Zakaj je pri medicinskih pripomočkih pristop drugačen kot pri zdravilih, kjer lekarne ne preverjajo posameznih certifikatov proizvajalcev?
Medicinski pripomočki in zdravila so urejeni z različnima zakonodajnima okviroma. Pri zdravilih je sistem dovoljenj za promet, sproščanja serij in nadzora dobavne verige urejen drugače kot pri medicinskih pripomočkih. Pri medicinskih pripomočkih pa MDR in IVDR izrecno določata obveznosti posameznih gospodarskih subjektov v dobavni verigi, vključno z obveznostmi distributerjev.
Zato lekarna pri medicinskih pripomočkih ne preverja dokumentacije zato, ker bi prevzemala vlogo proizvajalca, uvoznika ali priglašenega organa, temveč zato, ker kot distributer ne sme omogočati dostopnosti pripomočkov, za katere ugotovi ali utemeljeno domneva, da niso skladni. Obseg preverjanja mora biti sorazmeren, organiziran v okviru internih postopkov in dokumentiran tako, da lekarna lahko izkaže potrebno skrbnost. Razmejene so odgovornosti kot tudi kazenske določbe v primeru nespoštovanja zahtev MDR/IVDR.
14. Kako postopajo lekarne v primeru prodaje brizg za odvisnike ali prodaje brizg staršem za doziranje zdravil otrokom?
Najprej je treba preveriti predvideni namen in navodila proizvajalca za konkretno brizgo oziroma pripomoček. Če se pripomoček omogoča nestrokovnjakom, mora subjekt zagotoviti ustrezno svetovanje in informacije za varno uporabo.
15. Pri komu zdravstvene ustanove oziroma zdravstveni delavci dobijo izjave o skladnosti in EC certifikate, če imajo distributerji uvedeno metodo vzorčenja in nimajo vseh DoC in EC certifikatov?
Zdravstvena ustanova oziroma zdravstveni delavec lahko dokumentacijo o skladnosti zahteva od subjekta, ki mu je pripomoček dobavil.
Distributer mora pred omogočanjem dostopnosti pripomočka preveriti zahteve iz 14. člena MDR, pri čemer lahko za določena preverjanja uporabi reprezentativno metodo vzorčenja. To ne pomeni, da dokumentacije ni mogoče pridobiti, temveč da distributer določenih preverjanj ne izvaja nujno za vsak posamezen pripomoček oziroma vsako enoto.
Če distributer z dokumentacijo ne razpolaga neposredno, jo lahko pridobi oziroma zagotovi prek uvoznika, pooblaščenega predstavnika ali proizvajalca. Uvoznik mora v skladu s 13. členom MDR hraniti izvod izjave EU o skladnosti in, kadar je ustrezno, izvod certifikata.
Pri tem je treba razlikovati med dokumentacijo, ki jo gospodarski subjekti hranijo oziroma zagotavljajo za dokazovanje skladnosti, in dokumentacijo oziroma dokazili, ki jih zdravstvena ustanova ali naročnik zahteva v okviru svojih notranjih postopkov.
16. Zakaj se zahteva, da zdravstveni delavci shranjujejo izjave o skladnosti in EC certifikate za vse prejete medicinske pripomočke, če bodo dokumenti v EUDAMED?
MDR in ZMedPri-1 ne določata splošne obveznosti, da bi morali zdravstveni delavci oziroma zdravstvene ustanove shranjevati izjave EU o skladnosti in certifikate za vse prejete medicinske pripomočke.
Obveznosti zdravstvenih ustanov se nanašajo predvsem na sledljivost in vodenje evidenc za določene pripomočke. V skladu s 27. členom MDR morajo zdravstvene ustanove, po možnosti v elektronski obliki, shraniti in hraniti UDI za pripomočke za vsaditev razreda III, ki so jim bili dobavljeni ali so jih dobavile. ZMedPri-1 v 12. členu določa, da zdravstvene ustanove na enak način vodijo evidenco tudi za pripomočke, ki jim je priglašeni organ izdal certifikat in jih imajo v lasti.
EUDAMED je namenjen večji preglednosti in dostopu do podatkov o pripomočkih, gospodarskih subjektih in certifikatih, vendar ne nadomešča obveznosti zdravstvenih ustanov glede vodenja evidenc in zagotavljanja sledljivosti. V evidenci je zato smiselno zagotoviti podatke, ki omogočajo identifikacijo in sledljivost pripomočka, vključno s podatkom o certifikatu priglašenega organa, kadar je to relevantno.
Hrambe kopij vseh izjav EU o skladnosti in certifikatov za vse prejete medicinske pripomočke MDR oziroma ZMedPri-1 ne določata kot splošne obveznosti zdravstvenih ustanov. Dokumentacijo o skladnosti lahko zdravstvena ustanova, kadar jo potrebuje za preverjanje, notranje postopke, razpisne postopke ali obravnavo zapletov, pridobi od subjekta, ki ji je pripomoček dobavil oziroma omogočil njegovo dostopnost.
17. Kartica o vsadku ne vsebuje podatka, ali je na primer mogoča preiskava z MR. Ali bo dopolnjena?
Kartica o vsadku in informacije, ki jih proizvajalec zagotovi pacientu z vsajenim pripomočkom, morajo vsebovati podatke iz 18. člena MDR. Ti vključujejo tudi opozorila, previdnostne ukrepe ali ukrepe glede razumno predvidljivih zunanjih vplivov, medicinskih pregledov ali okoljskih razmer, kadar so za konkretni pripomoček relevantni.
Če je za varno uporabo vsadka pomembna informacija glede MR preiskave, mora biti ta zagotovljena v informacijah proizvajalca za pacienta oziroma v priloženih informacijah o pripomočku. Ni pa nujno, da so vse takšne informacije navedene neposredno na sami kartici o vsadku, če so pacientu zagotovljene na drug ustrezen način skupaj s kartico.
Zdravstvena ustanova mora v skladu z ZMedPri-1 pacienta seznaniti z informacijami iz 18. člena MDR, mu izročiti kartico o vsadku in podatke o vsajenih pripomočkih poslati v CeZZ. Dodatna pojasnila glede kartice o vsadku so podana v smernici MDCG 2019-8 v2.
18. Kartica o vsadku ne vsebuje vseh informacij za kasnejše zdravstvene obravnave. Ali boste predlagali dopolnitve?
Vsebina kartice o vsadku in informacij, ki jih mora proizvajalec zagotoviti pacientu z vsajenim pripomočkom, je določena z 18. členom MDR. Proizvajalec mora zagotoviti predpisane informacije, zdravstvena ustanova pa mora pacienta z njimi seznaniti in mu izročiti kartico o vsadku.
ZMedPri-1 v 11. členu dodatno določa obveznosti zdravstvenih ustanov glede posredovanja podatkov o vsajenih pripomočkih. Kartica o vsadku in podatki o vsajenih pripomočkih vključujejo tudi podatke o zdravstveni ustanovi, ki je pripomoček vsadila oziroma pacienta obravnavala, kot so ime in naslov zdravstvene ustanove ter njeni kontaktni podatki.
Če se v praksi pokaže potreba po dodatnih informacijah za kasnejše zdravstvene obravnave, se lahko te potrebe obravnavajo v okviru evropskih razprav, smernic ali morebitnih sprememb zakonodaje. JAZMP ne more samostojno spremeniti zahtev, ki jih glede vsebine kartice o vsadku določa MDR.
Dodatne informacije so lahko pacientu oziroma zdravstvenim delavcem zagotovljene tudi v drugih informacijah proizvajalca, če so potrebne za varno uporabo oziroma nadaljnjo obravnavo pacienta.
19. Precej medicinskih pripomočkov za profesionalno rabo ne gre v breme ZZZS in jih ni mogoče predpisati na zdravniško naročilnico. Kdaj bo urejena t. i. bela naročilnica?
Ureditev naročilnic, pravic iz obveznega zdravstvenega zavarovanja in morebitnih samoplačniških naročilnic ni v pristojnosti JAZMP. Z vidika ZMedPri-1 pa je pomembno, da se pripomočki, ki jih proizvajalec opredeli za profesionalno uporabo, nestrokovnjakom omogočajo le pod pogoji, ki jih določa zakon.
H. Optiki
1. Ali je mogoče pri optikah urediti poseben zavihek oziroma ločeno pojasnilo, ker je iz členov težko izločiti, katere zakonodaje se morajo držati?
Optiki so v skladu z MDR/IVDR opredeljeni kot distributerji in morajo spoštovati določbe 14. člena MDR/IVDR ali kot uvozniki, v kolikor pripomoček pridobijo iz tretjih držav. V tem primeru morajo spoštovati določbe 13. člena MDR/IVDR ter na pripomočku navesti informacije v skladu s členom 13(3).
2. Kakšne so zahteve za optične leče?
Optične leče za korekcijo vida so praviloma medicinski pripomočki razreda I. Optik mora najprej ugotoviti, v kateri vlogi nastopa: najpogosteje kot distributer, lahko pa tudi kot uvoznik ali v posebnih primerih kot subjekt z obveznostmi proizvajalca. Pri omogočanju dostopnosti optičnih leč mora preveriti, da so pripomočki skladni z MDR, označeni s CE, da je zanje pripravljena izjava EU o skladnosti, da so zagotovljene ustrezne informacije za uporabnika v slovenskem jeziku ter da je zagotovljena sledljivost v dobavni verigi. Če optik izdelek trži pod svojim imenom, spremeni njegov predvideni namen ali poseže v označevanje oziroma embalažo na način, ki vpliva na skladnost, se lahko uporabijo dodatne obveznosti po MDR, ki izhajajo iz 16. člena MDR in ZMedPri-1,
3. Proizvajalec za tekočino za nego kontaktnih leč nima avtorizacije za prodajo v Sloveniji, optik pa istega izdelka ne more dobaviti. Hkrati ga lahko potrošnik kupi na Amazonu ali drugi spletni platformi. Kako je to mogoče?
Dostopnost izdelka na spletni platformi še ne pomeni nujno, da je dobavna pot za slovenski trg urejena skladno z MDR/IVDR in ZMedPri-1. Slovenski optik kot gospodarski subjekt ne sme omogočiti dostopnosti pripomočka, če ne more zagotoviti skladnosti, ustreznih informacij, dobavne verige in izpolnitve svojih obveznosti. Pri spletnih platformah iz drugih držav se nadzor izvaja v okviru pristojnosti organov države sedeža subjekta, sodelovanja med organi in nadzora trga. Za medicinske pripomočke znotraj EU velja prost pretok blaga. To pomeni, da se pripomoček lahko trži v Uniji, v kolikor ima proizvajalec ali njegov pooblaščeni predstavnik sedež v eni izmed držav članic EU.
4. Optiki smo kot distributerji dolžni skrbeti za skladnost kontaktnih leč, tudi barvnih. Pri očalih pa izdelujemo očala za posameznega uporabnika. Kako to vpliva na pristojbino in ali potrebujemo mnenje revizorja?
Optiki pri kontaktnih lečah, vključno z barvnimi kontaktnimi lečami, ki so pripomočki brez medicinskega namena iz Priloge XVI in morajo biti prav tako skladni z MDR, praviloma nastopajo kot distributerji. Pri korekcijskih očalih pa praviloma ne gre za pripomočke, izdelane za posameznega uporabnika, saj so očala, sestavljena iz okvirja in stekel ter prilagojena uporabniku v skladu z navodili proizvajalca, največkrat prilagojeni pripomočki in ne pripomočki, izdelani za posameznega uporabnika. Za pristojbino je zato odločilno, v kateri vlogi subjekt nastopa po MDR, IVDR, ZMedPri-1 in Tarifi JAZMP. Če optik nastopa kot distributer, se pristojbina določi po pravilih za distributerje. Če vodi ustrezno ločeno računovodstvo in ločene računovodske evidence, je lahko osnova za pristojbino čisti letni prihodek, ustvarjen s prometom s pripomočki. Če želi tak ločeno izkazan promet uveljavljati pri JAZMP, mora do 30. septembra tekočega leta posredovati mnenje revizorja, kot je opredeljeno v 4. členu Tarife JAZMP za področje medicinskih pripomočkov.
I. Tarifa JAZMP za področje medicinskih pripomočkov
1. Kdo ureja oziroma določa višino tarif za distributerje? Je to določeno na ravni EU ali Slovenije? Od kod logika izračuna od prometa in zakaj ne od dobička?
Tarifa JAZMP za področje medicinskih pripomočkov je nacionalni akt Republike Slovenije, sprejet in objavljen po predpisanem postopku. JAZMP pri odmeri pristojbin uporablja veljavno objavljeno tarifo.
MDR in IVDR določata obveznosti tako gospodarskih subjektov kot pristojnih organov. V111. členu MDR in 104. členu IVDR, je navedeno, da uredbi ne posegata v možnost držav članic, da zaračunavajo pristojbine za dejavnosti, če se višina pristojbin določi na pregleden način in na podlagi načel povračila stroškov.
V času priprave Tarife JAZMP za področje medicinskih pripomočkov je bil izveden sestanek na Ministrstvu za zdravje, kjer so sodelovali predstavniki Ekonomsko socialnega sveta, Obrtna zbornica Slovenije, Trgovinska zbornica Slovenije in Medtech Slovenija. Po izvedenem sestanku se je višina pristojbin znižala, uredila so se razmerja med posameznimi poslovnimi subjekti in pripravljenih je bilo več plačilnih razredov.
Hkrati je bila v 4. člen Tarife JAZMP za področje pripomočkov dodana dodatna olajšava in sicer:
»Distributerji za plačilo letnih pristojbin s čistim prihodkom od prodaje manj od 350.000 eurov predložijo mnenje revizorja izdano v skladu s standardi revidiranja in pravili revidiranja na vsaka tri leta.« Veljavna tarifa tako določa odmerne razrede in osnovo za odmero pristojbin.
2. Ali bo zahtevke za plačilo tarife vsakemu deležniku poslala JAZMP s priloženim izračunom? Kdaj je predviden prvi zahtevek za plačilo?
JAZMP zavezancu pošlje poziv za plačilo letne pristojbine na elektronski naslov, ki ga je zavezanec navedel ob vpisu v register poslovnih subjektov. Tarifa določa, da se letna pristojbina plača v 15 dneh po prejemu poziva. Za zavezance, ki so na dan 1. januar 2026 že vpisani v register poslovnih subjektov, JAZMP poziv pošlje najpozneje do 30. junija tekočega koledarskega leta.
Za leto začetka veljavnosti tarife se uporablja prehodna oziroma sorazmerna odmera v skladu s tarifo. Pri subjektu, ki se prvič vpiše v register, je osnova za letno pristojbino najnižji znesek iz odmerne tabele za dejavnost, ki jo opravlja; če se registrira po 1. juliju tekočega leta, plača 50 % osnove za pristojbino. Tarifa nadalje določa, da se letna pristojbina v letu, ko sprejeta tarifa začne veljati, odmeri v sorazmernem deležu glede na število mesecev do konca koledarskega leta. Tarifa je bila v Uradnem listu RS objavljena 30. 1. 2026 in stopila v uporabo naslednji dan po objavi.
3. V Tarifi je določeno ločeno računovodstvo za medicinske pripomočke in mnenje revizorja. Na kakšni podlagi se zahteva mnenje revizorja kot pogoj pri izračunu pristojbine?
Tarifa določa, da poslovni subjekt, ki kot osnovo za določitev letne pristojbine uveljavlja ločeno računovodsko izkazovanje dejavnosti s pripomočki, vodi ločene računovodske evidence tako, da je jasno razlikovanje med dejavnostjo s pripomočki in drugo dejavnostjo. V tem primeru mora JAZMP najpozneje do 30. septembra tekočega leta posredovati mnenje revizorja, izdano v skladu s standardi in pravili revidiranja, s katerim izkazuje ustvarjeni promet s pripomočki. Na tej podlagi se izvede poračun letne pristojbine.
JAZMP pri odmeri uporablja veljavno tarifo. Vprašanja pravne ureditve oziroma morebitne spremembe te zahteve se lahko obravnavajo le v okviru postopka spremembe tarife oziroma pri pristojnih organih.
4. Zakaj je potrebno revizorsko poročilo tudi za tiste, ki sicer niso zavezani reviziji?
Mnenje revizorja je povezano z dokazovanjem ločenega računovodskega izkazovanja prometa s pripomočki, kadar subjekt želi, da se ta promet uporabi pri odmeri oziroma poračunu pristojbine in mora biti izdano v skladu s standardi in pravilih revidiranja. Če subjekt takega ločenega prometa ne uveljavlja, se uporabi osnova, določena v tarifi, praviloma podatki iz letnih poročil pri AJPES. Za distributerje s čistim prihodkom od prodaje manj kot 350.000 eurov tarifa določa, da mnenje revizorja predložijo na vsaka tri leta.
5. Smo podjetje, kjer nimamo mnenja revizorja. Kako do tega mnenja, ki bo osnova za izračun nadomestila JAZMP?
Subjekt mora vzpostaviti ločeno računovodsko izkazovanje prometa s pripomočki in pridobiti mnenje revizorja v skladu s standardi in pravili revidiranja, kot določa tarifa. JAZMP ne določa konkretnega revizorja. Če subjekt mnenja ne predloži oziroma ne izkaže ločenega prometa na način, ki ga določa tarifa, se pristojbina odmeri na podlagi razpoložljivih podatkov iz letnih poročil pri AJPES oziroma po pravilih tarife.
6. Zanima me logika tako izrazito degresivne lestvice pristojbin. Ali je regulator razmišljal o bolj proporcionalnem modelu?
Vprašanje razmerij med odmernimi razredi in modela odmere je vprašanje sprejete tarife. JAZMP pri izvajanju odmere uporablja veljavno tarifo, kot je bila sprejeta in objavljena. Morebitna sprememba degresivnosti, razredov ali načina izračuna bi bila mogoča le s spremembo tarife po predpisanem postopku.
7. Plačaš tudi, če nimaš nič prometa?
Obveznost plačila letne pristojbine se presoja glede na to, ali je subjekt zavezanec po tarifi in ali je registriran oziroma opravlja dejavnost, za katero tarifa določa letno pristojbino. Odmerne tabele vključujejo tudi najnižji razred. Zato ničelni ali zelo nizek promet sam po sebi ne pomeni nujno, da obveznosti plačila ni. Če subjekt dejavnosti ne opravlja več ali zanj ne obstaja več obveznost registracije, mora urediti spremembo oziroma izbris v skladu z ZMedPri-1.
8. Glede na to, da informacijski sistem še ne deluje in da so tarife namenjene tudi vzdrževanju sistema, ali bi se morale tarife za letos oziroma do delovanja sistema zmanjšati?
JAZMP pri odmeri uporablja veljavno tarifo. Za leto 2026 bo pristojbina odmerjena sorazmerno, in sicer od najnižjega zneska iz ustrezne odmerne tabele.
Naloge JAZMP se izvajajo tudi v prehodnem obdobju pred polno vzpostavitvijo informacijskega sistema.
9. Koliko znaša letna pristojbina, če letni promet z medicinskimi pripomočki znaša le 10 % celotnega prometa?
Tarifa za distributerje, uvoznike in subjekte, ki so hkrati distributerji in uvozniki, kot osnovo praviloma uporablja čisti letni prihodek od prodaje iz razpoložljivih podatkov iz letnih poročil pri AJPES. Na podlagi te osnove se subjekt uvrsti v ustrezni razred odmerne tabele.
Če želi subjekt, da se pri odmeri oziroma poračunu upošteva samo promet z medicinskimi pripomočki, mora ta promet ločeno računovodsko izkazovati in predložiti mnenje revizorja v skladu s tarifo. V tem primeru se subjekt v odmerno tabelo uvrsti glede na izkazani promet z medicinskimi pripomočki.
10. Če smo registrirani samo za pripomočke iz najnižjega razreda ali se lahko izpišemo iz registra? Kako je v tem primeru s pristojbino?
Če subjekt omogoča dostopnost izključno pripomočkov, za katere ZMedPri-1 določa izjemo od obveznosti registracije distributerja, mora preveriti, ali zanj obveznost registracije še obstaja. Če pogoji za registracijo niso več podani ali subjekt preneha opravljati dejavnost, mora spremembo oziroma izbris urediti pri JAZMP. Obveznost plačila pristojbine se presoja glede na status zavezanca in podatke v registru v relevantnem obdobju.
11. Lekarnam ZZZS nameni manj kot 20 % marže na medicinske pripomočke. Kako se to upošteva pri tarifi?
Vprašanja marž, financiranja in pravil ZZZS niso predmet Tarife JAZMP in niso v pristojnosti JAZMP.
