1. Kaj je medicinski pripomoček?
„medicinski pripomoček“ pomeni vsak instrument, aparat, napravo, programsko opremo, vsadek, reagent, material ali drug predmet, za katerega je proizvajalec predvidel, da se uporablja samostojno ali v kombinaciji za ljudi za enega ali več naslednjih specifičnih medicinskih namenov:
- diagnosticiranje, preventivo, spremljanje, predvidevanje, prognozo, zdravljenje ali lajšanje bolezni,
- diagnosticiranje, spremljanje, zdravljenje, lajšanje poškodb ali invalidnosti ali nadomestilo zanje,
- preiskovanje, nadomeščanje ali spreminjanje anatomije ali fiziološkega ali patološkega procesa ali stanja,
- pridobivanje informacij z in vitro preiskavami vzorcev, pridobljenih iz človeškega telesa, vključno z darovanimi organi, krvjo in tkivi,
in ki svojega glavnega predvidenega učinka na človeško telo ne dosega na farmakološki, imunološki ali metabolični način, vendar so mu ti procesi v pomoč pri njegovem delovanju.
2. Kaj še sodi med medicinske pripomočke?
Med medicinske pripomočke sodijo tudi:
- pripomočki za nadziranje spočetja ali pomoč pri njem,
- izdelki, posebej namenjeni čiščenju, razkuževanju ali sterilizaciji pripomočkov,
- dodatki za medicinske pripomočke,
- izdelki iz Priloge XVI (izdelki brez predvidenega medicinskega namena).
3. Kaj je dodatek za medicinski pripomoček?
„dodatek za medicinski pripomoček“ pomeni predmet, ki sam sicer ni medicinski pripomoček, vendar ga je njegov proizvajalec namenil za uporabo skupaj z enim ali več posebnimi medicinskimi pripomočki, in izrecno omogoča uporabo medicinskega pripomočka ali pripomočkov v skladu z njihovimi predvidenimi nameni ali izrecno in neposredno pripomore k medicinski funkcionalnosti medicinskega pripomočka ali pripomočkov z vidika njihovega predvidenega namena.
4. Kaj je in vitro diagnostični medicinski pripomoček?
In vitro diagnostični medicinski pripomoček je skladno z definicijo iz IVDR vsak medicinski pripomoček, ki je reagent, reagenčni izdelek, umerjevalec, kontrolni material, komplet, instrument, aparat, del opreme, programska oprema ali sistem, ki se uporablja sam ali v kombinaciji, proizvajalec pa ga je predvidel za uporabo in vitro za preiskave vzorcev, vključno z darovano krvjo in darovanimi tkivi, pridobljenimi iz človeškega telesa, samo ali v glavnem za pridobivanje ene ali več naslednjih informacij:
- o fiziološkem ali patološkem procesu ali stanju;
- o prirojenih telesnih ali duševnih prizadetostih;
- o nagnjenosti k bolezenskemu stanju ali bolezni;
- za določanje varnosti in združljivosti z možnimi prejemniki;
- za predvidevanje odzivanja ali reakcij na zdravljenje;
- za opredelitev ali spremljanje terapevtskih ukrepov.
Za in vitro diagnostične medicinske pripomočke se štejejo tudi posode za vzorce, ki so pripomočki, vakuumski ali ne, ki jih je proizvajalec namenil predvsem za to, da vsebujejo in hranijo vzorce, vzete iz človeškega telesa za namene in vitro diagnostične preiskave.
5. Kaj so dodatki za in vitro diagnostične medicinske pripomočke?
IVDR ureja tudi dodatke za in vitro diagnostične medicinske pripomočke, pripomočke za samo-testiranje, pripomočke za testiranje ob pacientu in dopolnilno diagnostiko.
Dodatek za in vitro diagnostični medicinski pripomoček pomeni predmet, ki sam sicer ni in vitro diagnostični medicinski pripomoček, vendar ga je njegov proizvajalec namenil za uporabo skupaj z enim ali več posebnimi in vitro diagnostičnimi medicinskimi pripomočki, in izrecno omogoča uporabo in vitro diagnostičnega medicinskega pripomočka ali pripomočkov v skladu z njihovimi predvidenimi nameni ali izrecno in neposredno pripomore k medicinski funkcionalnosti in vitro diagnostičnega medicinskega pripomočka ali pripomočkov z vidika njihovega predvidenega namena.
6. Kaj je pripomoček za samo-testiranje?
Pripomoček za samo-testiranje pomeni vsak pripomoček, ki ga je proizvajalec namenil za uporabo nestrokovnjakom, vključno s pripomočki, ki se uporabljajo za storitve testiranja, ki se ponujajo nestrokovnjakom v okviru storitev informacijske družbe.
Pripomoček za testiranje ob pacientu pomeni vsak pripomoček, ki ni namenjen za samo-testiranje, temveč za testiranje zunaj laboratorijskega okolja, ki ga izvaja zdravstveni delavec, običajno blizu pacienta ali ob njem.
7. Kaj pomeni dopolnilna diagnostika?
Dopolnilna diagnostika pomeni pripomoček, ki je bistven za varno in učinkovito uporabo ustreznega zdravila, da se:
- pred in/ali med zdravljenjem ugotovi, za katere paciente je najbolj verjetno, da jim bo ustrezno zdravilo koristilo, ali
- pred in/ali med zdravljenjem ugotovi, pri katerih pacientih verjetno obstaja povečano tveganje resnih neželenih učinkov zaradi zdravljenja z ustreznim zdravilom.
8. Katera zakonodaja ureja področje pripomočkov?
Leta 2017 sta bili na nivoju EU sprejeti dve uredbi:
- Uredba (EU) 2017/745 Evropskega parlamenta in sveta o medicinskih pripomočkih – MDR
- Uredba (EU) 2017/746 o in vitro diagnostičnih medicinskih pripomočkih – IVDR
Začetek uporabe:
MDR: 26.5.2021
IVDR: 26.5.2022
Veliko informacij je objavljenih na spletni strani Evropske Komisije. Na isti strani pod zavihkom GUIDANCE najdete zbrane smernice, sprejete s strani Koordinacijske skupine za medicinske pripomočke (MDCG), ki deležnikom pomagajo pri implementaciji določil uredb.
9. Kako je z nacionalno zakonodajo?
Od 19. junija 2025 je v veljavi Zakon o medicinskih pripomočkih (ZMedPri-1, Uradni list RS, št. 40/25), ki ureja področje medicinskih pripomočkov, dodatkov za medicinske pripomočke, pripomočkov brez medicinskega namena ter in vitro diagnostičnih medicinskih pripomočkov.
ZMedPri-1 na nacionalni ravni ureja pristojne organe za izvajanje Uredbe 2017/745/EU, Uredbe 2017/746/EU in Uredbe 2022/123/EU, jezik, ponovno obdelavo in nadaljnjo uporabo pripomočkov za enkratno uporabo, vzpostavitev informacijskega sistema JAZMP, pogoje za distributerje medicinskih pripomočkov, izredno odobritev dajanja pripomočkov na trg, informacije za paciente, ki jim je bil vsajen pripomoček, ter informacije, ki jih hranijo zdravstvene ustanove, omogočanje pošiljanja informacij nestrokovnjakom, oglaševanje, registracijo poslovnih subjektov in pripomočkov, klinične raziskave in študije učinkovitosti ter sodelovanje med Komisijo za medicinsko etiko in JAZMP, vigilanco, pristojbine ter prekrške in sankcije za kršitve določb Uredbe 2017/745/EU, Uredbe 2017/746/EU, Uredbe 2022/123/EU in tega zakona.
10. Kakšne so prehodne določbe MDR in IVDR in kakšna je veljavnost certifikatov?
Informacije so dostopne na povezavi:
https://www.jazmp.si/medicinski-pripomocki/splosno-o-medicinskih-pripomockih/novi-uredbi-mdr-in-ivdr/
11. Kaj pomeni pripomoček izdelan za posameznega uporabnika?
„pripomoček, izdelan za posameznega uporabnika“ pomeni vsak pripomoček, posebej izdelan v skladu z naročilnico katere koli osebe, ki je po nacionalnem pravu pooblaščena zaradi svojih poklicnih kvalifikacij, v kateri so na odgovornost te osebe navedene posebne značilnosti zasnove, in ki je namenjen samo uporabi pri določenem pacientu in izključno za zadovoljitev njegovega osebnega stanja in potreb.
Vendar se masovno proizvedeni pripomočki, ki jih je treba prilagoditi, da bi ustrezali posebnim zahtevam vseh poklicnih uporabnikov, in pripomočki, ki so masovno izdelani z industrijskimi postopki izdelave v skladu z naročilnicami katere koli pooblaščene osebe, ne štejejo za pripomočke, izdelane za posameznega uporabnika.
Povezava na smernico MDCG 2021-3 – ”Questions and answer on Custom-Made Devices”:
https://health.ec.europa.eu/system/files/2021-03/mdcg_2021-3_en_0.pdf
12. Kaj pomeni aktivni pripomoček?
„aktivni pripomoček“ pomeni vsak pripomoček, katerega delovanje je odvisno od vira energije, ki je v ta namen ne tvori človeško telo ali gravitacija, in ki deluje s spreminjanjem gostote ali pretvarjanjem te energije. Pripomočki, ki so namenjeni prenosu energije, snovi ali drugih elementov med aktivnim pripomočkom in pacientom brez konkretne spremembe, se ne štejejo za aktivne pripomočke.
Tudi programska oprema se šteje za aktivni pripomoček.
13. Kaj pomeni pripomoček za vsaditev?
„pripomoček za vsaditev“ pomeni vsak pripomoček, vključno s tistimi, ki se delno ali v celoti absorbirajo, ki je namenjen: — popolni vstavitvi v človeško telo ali — zamenjavi epitelne površine ali površine očesa s kliničnim posegom, pri čemer po opravljenem postopku ostane v telesu. Za pripomoček za vsaditev se šteje tudi vsak pripomoček, ki je namenjen delni vstavitvi v človeško telo s kliničnim posegom in ki po opravljenem postopku ostane v telesu najmanj 30 dni.
14. Kakšna je vloga JAZMP na področju medicinskih pripomočkov?
- vodi registre poslovnih subjektov s sedežem v Republiki Sloveniji, ki opravljajo dejavnost na področju medicinskih pripomočkov;
- vodi Register medicinskih pripomočkov proizvajalcev ter pooblaščenih predstavnikov s sedežem v Republiki Sloveniji;
- izdaja Certifikate o prosti prodaji;
- vodi postopke potrditve in ocenjevanja vlog za klinične raziskave in študije učinkovitosti;
- vodi postopke na vigilančnem področju in mesečno na spletni strani objavlja prejeta obvestila o varnostnih korektivnih ukrepih;
- vodi seznam pripomočkov, katerih dostopnost lahko poslovni subjekti omogočajo brez strokovnega svetovanja;
- obravnava obvestila o prenehanju ali prekinitvi dobave pripomočkov, skladno s točko (a) 10. člena MDR in IVDR, uvedeno z Uredbo 2024/1860/EU;
- izvaja nadzor (nad izvajanjem določb Uredbe 2017/745/EU, Uredbe 2017/746/EU, Uredbe 2022/123/EU in zakona; npr. preverjanje skladnosti pripomočkov na trgu RS, izpolnjevanja pogojev in obveznosti slovenskih gospodarskih subjektov);
- vodi postopke izrednih odobritev za dajanje na trg ali v uporabo (59. člen MDR in 54. člen IVDR);
- imenuje in nadzoruje priglašene organe v RS;
- sodeluje v mednarodnih EU koordinacijskih in delovnih skupinah;
- sodeluje v krovnih skupinah EU kot so: MDCG, CAMD, HMA Core Group, Medical Devices Committe.
15. Katere gospodarske subjekte definirata uredbi?
„proizvajalec“ pomeni fizično ali pravno osebo, ki izdeluje ali popolnoma predela pripomoček ali naroči zasnovo, izdelavo ali popolno predelavo pripomočka in ki ta pripomoček trži pod svojim imenom ali blagovno znamko,
„pooblaščeni predstavnik proizvajalca“ pomeni vsako fizično ali pravno osebo s sedežem v Uniji, ki je prejela in sprejela pisno pooblastilo proizvajalca, ki se nahaja zunaj Unije, da v njegovem imenu izvaja določene naloge v zvezi z obveznostmi tega proizvajalca po uredbi,
„uvoznik“ pomeni vsako fizično ali pravno osebo s sedežem v Uniji, ki daje pripomoček iz tretje države na trg Unije,
„distributer“ pomeni vsako fizično ali pravno osebo v dobavni verigi, ki ni proizvajalec ali uvoznik in ki omogoča dostopnost pripomočka na trgu, dokler ta ni dan v uporabo,
„proizvajalec sistema ali paketa“ v skladu z 22. členom MDR – pripomočki z oznako CE se kombinirajo z drugimi pripomočki ali izdelki na način, ki je skladen s predvidenim namenom in v mejah uporabe, ki jo določi proizvajalec pripomočka (priprava izjave),
„zdravstvena ustanova“ organizacija, katere glavni namen je nega ali zdravljenje pacientov ali promocija javnega zdravja.
16. Kdo je odgovoren za razvrstitev izdelka med medicinske pripomočke in njegovo razvrstitev?
Za razvrstitev izdelka med medicinske pripomočke in njegovo razvrstitev je odgovoren proizvajalec.
V skladu z Uredbo 2017/745/EU so pripomočki razvrščeni v različne stopnje tveganja za uporabnika. Za najnižji razred tveganja, razred I, proizvajalec pripravi Izjavo o skladnosti, s katero zagotavlja, da je pripomoček varen, skladen in učinkovit. Za ostale razrede tveganja pripomočka (razred I – sterilno, I – z merilno funkcijo, IIa, IIb in III) mora proizvajalec v presojo skladnosti vključiti priglašeni organ, ki mu po končani presoji sledi izdaja certifikata EU. Tak pripomoček ima poleg CE oznake tudi štirimestno številko priglašenega organa, ki je ocenjevanje opravil.

Z Uredbo 2017/746/EU se popolnoma spreminja klasifikacija in vitro diagnostičnih medicinskih pripomočkov in jih deli v razrede tveganja: A, A sterilno, B, C, D. Za najnižji razred tveganja, razred A, proizvajalec pripravi Izjavo o skladnosti, s katero zagotavlja, da je pripomoček varen, skladen in učinkovit. Za ostale razrede tveganja pripomočka (razred A – sterilno, B, C in D) mora proizvajalec v presojo skladnosti vključiti priglašeni organ, ki mu po končani presoji sledi izdaja certifikata EU. Tudi tak pripomoček ima poleg CE oznake tudi štirimestno številko priglašenega organa, ki je ocenjevanje opravil.
Ob tem je pomembno tudi, da bo za zadostitev zahtev Uredbe 2017/746/EU večina in vitro diagnostičnih medicinskih pripomočkov v presojo skladnosti potrebovala vključitev priglašenega organa in posledično izdan EU certifikat. Na sliki spodaj je razvidna sprememba klasifikacije:

17. Kakšne so zakonske obveznosti poslovnih/gospodarski subjektov?
Registracija poslovnega/gospodarskega subjekta v registre dejavnosti: Poslovni/gospodarski subjekti s sedežem v Republiki Sloveniji, ki opravljajo dejavnost povezano z medicinskimi pripomočki, kot je njihovo dajanje na trg ali v uporabo, omogočanje njihove dostopnosti na trgu ali pa so zanje pooblaščeni za zastopanje na trgu Unije s strani proizvajalca iz tretje države, se morajo pred začetkom izvajanja dejavnosti registrirati pri JAZMP.
Registracija medicinskega pripomočka v Register pripomočkov v RS: Obveznost registracije medicinskega pripomočka ima proizvajalec s sedežem v Republiki Sloveniji in pooblaščeni predstavnik s sedežem v Republiki Sloveniji, ki je imenovan s strani proizvajalca iz tretje države.
18. Kakšne so zakonske obveznosti proizvajalcev in pooblaščenih predstavnikov proizvajalca glede registracije medicinskih pripomočkov?
V skladu s prehodnimi določbami obeh uredb (tretji odstavek 123. člena MDR in tretji odstavek 113. člena IVDR) do uporabe podatkovne zbirke EUDAMED proizvajalec s sedežem v Republiki Sloveniji ali pooblaščeni predstavnik proizvajalca s sedežem v Republiki Sloveniji preden da pripomoček, ki ni pripomoček, izdelan za posameznega uporabnika, na trg na JAZMP predloži vlogo za registracijo pripomočka v skladu s 45. členom ZMedPri-1 (Uradni list RS, št. 40/25).
Navodila, pojasnila in dostop do obrazcev je na spletni strani JAZMP na povezavi: https://www.jazmp.si/medicinski-pripomocki/registracija-medicinskih-pripomockov/.
19. Definicije
»omogočanje dostopnosti« pomeni vsako dobavo pripomočka, razen pripomočka, ki je predmet raziskave, za distribucijo, potrošnjo ali uporabo na trgu Unije.
»dajanje na trg« pomeni prvo omogočanje dostopnosti pripomočka na trgu Unije, razen pripomočka, ki je predmet raziskave.
»dajanje v uporabo« pomeni fazo, ko je pripomoček, ki ni pripomoček, ki je predmet raziskave, prvič dostopen končnemu uporabniku na trgu Unije za predviden namen.
20. Kam sodijo specializirane prodajalne?
Uredbi ne ločujeta več med prodajo na debelo in drobno. Vse specializirane prodajalne prevzemajo odgovornosti bodisi distributerjev bodisi uvoznikov.
21. Kaj mora biti navedeno na embalaži pripomočka in kateri subjekti?
Natančna navodila glede označevanja pripomočka so navedena v Prilogi I (Splošne zahteve glede varnosti in učinkovitosti) obeh uredb.
Simboli na embalaži pripomočka morajo biti skladni s SIST EN ISO 15223-1:2021.
22. Kakšne so obveznosti distributerjev in kaj vse mora preveriti distributer pred omogočanjem dostopnosti pripomočka na trgu?
Obveznosti distributerjev so navedene v 14. členu MDR in 14. členu IVDR.
Distributerji pred omogočanjem dostopnosti pripomočka na trgu preverijo, ali so izpolnjene vse naslednje zahteve:
- je pripomoček opremljen z oznako CE in je zanj pripravljena Izjava EU o skladnosti ter v primeru višjega razreda tveganja izdan certifikat priglašenega organa
- so pripomočku priložene ustrezne informacije s strani proizvajalca (zahteve Priloge I točke 23 MDR),podatki na oznaki pa so neizbrisni, čitljivi in jasno razumljivi predvidenemu uporabniku ali pacientu,
- je uvoznik izpolnil zahteve iz člena 13(3) MDR – navedba imena uvoznika, kraj poslovanja itd.,
- proizvajalec je določil UDI, kjer je ustrezno.
Ostale obveznosti distributerjev:
- Zagotavljanje, da so v času, ko so odgovorni za pripomoček, pogoji skladiščenja ali prevoza skladni s pogoji, ki jih je določil proizvajalec.
- Obveščanje pristojnih organov (v RS JAZMP), kadar menijo ali domnevajo, da pripomoček pomeni resno tveganje, vključno z navedbo neskladnosti in morebitnih sprejetih korektivnih ukrepov.
- Sodelovanje s proizvajalcem, po potrebi tudi pooblaščenim predstavnikom in uvoznikom, ter pristojnimi organi ob domnevi, da pripomoček ni skladen.
- Posredujejo pritožbe ali poročila zdravstvenih delavcev, pacientov ali uporabnikov o sumih na zaplete proizvajalcu ter po potrebi pooblaščenemu predstavniku ali uvozniku.
- Vodenje registra pritožb, neskladnih, odpoklicanih in umaknjenih pripomočkov.
- Sodelovanje s pristojnim organom glede dokazovanja skladnosti pripomočka.
- Sodelovanje s pristojnimi organi na njihovo zahtevo pri vsaki dejavnosti za odpravo tveganj, povezanih s pripomočki, katerih dostopnost na trgu so omogočili. Na zahtevo pristojnega organa zagotovijo brezplačne vzorce pripomočka ali, kadar to ni izvedljivo, omogočijo dostop do pripomočka.
- Sodelujejo s proizvajalci ali pooblaščenimi predstavniki za zagotavljanje ustrezne ravni sledljivosti.
Obveznosti distributerjev so navedene tudi v Priročniku za distributerje, ki je objavljen na spletni strani JAZMP v zavihku Registracija poslovnih subjektov. Priročnik povzema zahteve Uredbe 2017/745/EU (MDR) in Uredbe 2017/745/EU (IVDR). Za izpolnitev zahtev nacionalne zakonodaje upoštevajte določila Zakona o medicinskih pripomočkih (ZMedPri-1, Uradni list RS, št. 40/25).
Smernica MDCG 2021-27 – ‘’Questions and Answers on Article 13 & 14 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746’’ dodatno pojasnjuje obveznosti uvoznikov in distributerjev.
23. Kakšne so obveznosti uvoznikov?
Obveznosti uvoznikov so navedene v 13. členu MDR in IVDR.
Uvozniki preden dajo pripomoček na trg, preverijo, da je:
- pripomoček opremljen z oznako CE ter da je pripravljena izjava EU o skladnosti pripomočka;
- proizvajalec znan in je imenoval pooblaščenega predstavnika v skladu s členom 11;
- pripomoček označen v skladu s to uredbo in so mu priložena potrebna navodila za uporabo;
- proizvajalec v skladu s členom 27 določil UDI, kjer je to ustrezno.
Ostale obveznosti uvoznikov:
- Uvozniki na pripomočku, njegovi embalaži ali spremnem dokumentu navedejo svoje ime, registrirano trgovsko ime ali registrirano blagovno znamko, svoj registrirani kraj poslovanja in naslov, na katerem so dosegljivi, tako da je mogoče ugotoviti naslov njihove dejanske lokacije.
- Zagotovijo, da dodatne oznake ne prekrivajo nobene informacije na oznaki proizvajalca.
- Zagotavljanje, da v času, ko so odgovorni za pripomoček, pogoji skladiščenja ali prevoza ne ogrožajo njegove skladnosti s splošnimi zahtevami glede varnosti in učinkovitosti, določenimi v Prilogi I, in da so izpolnjeni morebitni pogoji, ki jih je določil proizvajalec.
- Obveščanje pristojnih organov (v RS JAZMP), kadar menijo ali domnevajo, da pripomoček pomeni resno tveganje, vključno z navedbo neskladnosti in morebitnih sprejetih korektivnih ukrepov.
- Vodenje registra pritožb, neskladnih, odpoklicanih in umaknjenih pripomočkov.Proizvajalcu, pooblaščenemu predstavniku in distributerjem sporočijo vse informacije, ki jih ti zahtevajo, da bi lahko preiskali pritožbe.
- Sodelovanje s pristojnim organom glede dokazovanja skladnosti pripomočka.
- Sodelovanje s pristojnimi organi na njihovo zahtevo pri vsaki dejavnosti za odpravo ali – če to ni mogoče – zmanjšanje tveganj, povezanih s pripomočki, ki so jih dali na trgu. Na zahtevo pristojnega organa zagotovijo brezplačne vzorce pripomočka ali, kadar to ni izvedljivo, omogočijo dostop do pripomočka.
- Uvozniki v obdobju iz 10(8). člena hranijo izvod izjave EU o skladnosti in, kjer je potrebno, izvod ustreznega certifikata, vključno z morebitnimi spremembami in dopolnili, izdanimi v skladu s 56. členom.
- Sodelujejo s proizvajalci ali pooblaščenimi predstavniki za zagotavljanje ustrezne ravni sledljivosti.
Za izpolnitev zahtev nacionalne zakonodaje upoštevajte določila Zakona o medicinskih pripomočkih (ZMedPri-1, Uradni list RS, št. 40/25).
Smernica MDCG 2021-27 – ‘’Questions and Answers on Article 13 & 14 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746’’ dodatno pojasnjuje obveznosti uvoznikov in distributerjev.
24. V katerih primerih se obveznosti proizvajalcev uporabljajo za uvoznike, distributerje in druge osebe?
V skladu s 16. členom MDR distributer, uvoznik ali druga fizična ali pravna oseba prevzame obveznosti proizvajalcev, če stori kar koli od naslednjega:
(a) omogoči dostopnosti pripomočka na trgu pod svojim imenom, registriranim trgovskim imenom ali registrirano blagovno znamko, razen v primerih, ko distributer ali uvoznik sklene dogovor s proizvajalcem, v skladu s katerim je proizvajalec kot tak naveden na oznaki in je odgovoren za izpolnjevanje zahtev, ki jih ta uredba nalaga proizvajalcem;
(b) spremeni predvideni namena pripomočka, ki je že dan na trg ali v uporabo;
(c) spremeni pripomoček, ki je že dan na trg ali v uporabo tako, da to lahko vpliva na skladnost z veljavnimi zahtevami.
Prvi pododstavek se ne uporablja za osebe, ki se sicer ne štejejo za proizvajalca, kakor je opredeljen pri 30. točki 2. člena MDR, vendar za posameznega pacienta sestavljajo ali prilagajajo pripomoček, ki je že na trgu, ne da bi spreminjale njegov predvideni namen.
25. Kakšne obveznosti ima poslovni subjekt, ki daje na novo označen ali prepakiran pripomoček na trg?
Obveznosti so določene v četrtem odstavku 16. člena MDR:
Vsaj 28 dni, preden je na novo označen ali prepakiran pripomoček dostopen na trgu, distributerji ali uvozniki, ki izvajajo katere koli od dejavnosti iz točk (a) in (b) drugega odstavka 16. člena, obvestijo proizvajalca in pristojni organ države članice, v kateri nameravajo omogočiti dostopnost pripomočka, o nameri omogočiti dostopnost na novo označenega ali prepakiranega pripomočka ter proizvajalcu in pristojnemu organu na zahtevo predložijo vzorec ali maketo na novo označenega ali prepakiranega pripomočka, vključno z vsemi prevedenimi oznakami in navodili za uporabo. V istem obdobju 28 dni distributer ali uvoznik pristojnemu organu predloži certifikat, ki ga izda priglašeni organ, imenovan za vrsto pripomočkov, ki so predmet dejavnosti iz točk (a) in (b) odstavka 2, s katerimi potrjuje, da je sistem vodenja kakovosti, vzpostavljen s strani distributerja ali uvoznika, skladen z zahtevami iz odstavka 3.
Na strani EK je objavljena smernica MDCG 2021-26 – ”Q & A on repackaging & relabelling activities under Article 16 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746”.
26. Kako je v skladu z uredbama definirana Oseba odgovorna za skladnost z zakonodajo in kašne pogoje mora izpolnjevati?
Proizvajalci imajo v svoji organizaciji vsaj eno osebo, ki je odgovorna za skladnost z zakonodajo in ima potrebno strokovno znanje na področju medicinskih pripomočkov (15. člen Uredbe 2017/745/EU oz. Uredbe 2017/746/EU).
Potrebno strokovno znanje se dokaže z eno od naslednjih kvalifikacij:
(a) diploma, spričevalo ali drugo dokazilo o formalnih kvalifikacijah, ki se podeljuje ob zaključku univerzitetnega izobraževanja ali študijskega programa, ki ga zadevna država članica priznava za enakovrednega na področju prava, medicine, farmacije, inženirstva ali druge ustrezne znanstvene vede, ter vsaj eno leto delovnih izkušenj z zakonodajnimi zadevami ali s sistemi vodenja kakovosti, povezanimi z medicinskimi pripomočki;
(b) štiri leta poklicnih izkušenj z zakonodajnimi zadevami ali s sistemi vodenja kakovosti, povezanimi z medicinskimi pripomočki.
Brez poseganja v nacionalne določbe glede poklicnih kvalifikacij lahko proizvajalci pripomočkov, izdelanih za posameznega uporabnika, potrebno strokovno znanje iz prvega pododstavka dokazujejo z vsaj dvema letoma poklicnih izkušenj na ustreznem proizvodnem področju.
V skladu s 20. členom Zakona o medicinskih pripomočkih (ZMedPri-1) proizvajalci, pooblaščeni predstavniki proizvajalca, proizvajalci pripomočkov, izdelanih za posameznega uporabnika, ter proizvajalci sistema in paketa iz 22. člena Uredbe 2017/745/EU ob registraciji JAZMP predložijo dokazila, ki izkazujejo ustreznost kvalifikacij osebe, odgovorne za skladnost z zakonodajo.
Mikro in malim podjetjem v smislu Priporočila Komisije 2003/361/ES znotraj organizacije ni treba imeti osebe, odgovorne za skladnost z zakonodajo, vendar pa jim mora biti takšna oseba stalno in nepretrgoma na voljo.
27. Kakšne so odgovornosti Osebe odgovorne za skladnosti z zakonodajo?
Oseba, odgovorna za skladnost z zakonodajo (MDR in IVDR), je odgovorna vsaj za zagotavljanje, da:
(a) je skladnost pripomočkov ustrezno preverjena v skladu s sistemom vodenja kakovosti, v okviru katerega so bili pripomočki izdelani, preden se pripomoček sprosti;
(b) sta tehnična dokumentacija in izjava EU o skladnosti sestavljeni in se redno posodabljata;
(c) so obveznosti glede nadzora po dajanju na trg izpolnjene v skladu z 10. odstavkom 10. člena MDR in z 9. odstavkom 10. člena IVDR;
(d) so obveznosti poročanja iz členov od 87 do 91 MDR in členov 82 do 86 IVDR izpolnjene;
(e) je pri pripomočkih, ki so predmet raziskave, izdana izjava iz oddelka 4.1 poglavja II Priloge XV MDR;
(f) se v primeru pripomočkov za študijo učinkovitosti, ki so namenjeni uporabi v okviru intervencijskih ali drugih študij učinkovitosti, ki vključujejo tveganja za udeležence v študiji, izda izjava iz oddelka 4.1. Priloge XIV IVDR.
28. Kdo pripravi Izjavo EU o skladnosti?
Izjavo EU o skladnosti pripravi proizvajalec pripomočka v skladu s Prilogo IV (Izjava EU o skladnosti) obeh uredb. Na izjavi je potrebno navesti vse zahtevane elemente omenjene priloge. S pripravo izjave EU o skladnosti proizvajalec prevzame odgovornost za skladnost z zahtevami iz te uredbe in vso drugo zakonodajo Unije, ki se uporablja za pripomočke. Proizvajalec izjavo EU o skladnosti redno posodablja.
29. Kako je v skladu z uredbama opredeljen Edinstveni identifikator pripomočka, Osnovni UDI-DI in Sistem UDI-DI ter kakšne prednosti prinaša?
Informacije so dostopne na povezavi:
https://www.jazmp.si/medicinski-pripomocki/splosno-o-medicinskih-pripomockih/mdcg-smernice-za-medicinske-pripomocke/edinstveni-identifikator-pripomocka-udi/
30. Ali je potrebna namestitev UDI na embalažo pripomočka ob dajanju pripomočka na trg?
Za namestitev UDI na embalažo pripomočka je določeno prehodno obdobje kot prikazuje slika:

31. Kako lahko izgleda nosilec UDI na pripomočku?
Obliko nosilca UDI na pripomočku določi proizvajalec skupaj s subjektom izdajateljem UDI. Nosilec je lahko v več oblikah: linearna koda, QR koda, RFID ali matrix koda:




32. Kateri so subjekti izdajatelji UDI po uredbi?
Subjekti izdajatelji so navedeni v 12. odstavku 120. člena MDR. To so GS1, HIBCC, ICCBBA in IFA.
Vsak subjekt izdajatelj ima za izdajo UDI svoj edinstven način, ki so opisani na naslednji spletni strani.
33. Kakšne so obveznosti zdravstvenih ustanov?
V skladu z 11. členom Zakona o medicinskih pripomočkih (ZMedPri-1) pacienta, ki mu je bil vsajen pripomoček, v okviru pojasnilne dolžnosti, ustno ali pisno, seznanijo z informacijami iz prvega odstavka 18. člena Uredbe 2017/745/EU, mu izročijo kartico o vsadku in v Centralni register podatkov o pacientih v skladu z zakonom, ki ureja zbirke podatkov s področja zdravstvenega varstva, pošljejo podatke o vsajenih pripomočkih:
- identifikacijska številka pacienta v CRPP,
- osebno ime pacienta,
- številka zdravstvene ustanove, ime in naslov zdravstvene ustanove, kontaktni podatki ustanove,
- datum vsaditve pripomočka,
- informacije, ki omogočajo identifikacijo pripomočka, vključno z imenom pripomočka, serijsko številko, številko partije, UDI, modelom pripomočka ter imenom in naslovom proizvajalca ter njegovim spletnim mestom (podatki iz točke (a) 1. odstavka 18. člena Uredbe 2017/745/EU).
V primeru varnostnega tveganja v zvezi z vsajenimi pripomočki lahko JAZMP od zdravstvenih ustanov ali Nacionalnega inštituta za javno zdravje zahteva podatke o številu in vrsti posameznega pripomočka, ki je bil vsajen. Zdravstvene ustanove in Nacionalni inštitut za javno zdravje se morajo odzvati takoj, najpozneje pa v treh dneh od prejema zahteve JAZMP.
Zdravstvene ustanove shranijo in hranijo, po možnosti v elektronski obliki, UDI za pripomočke, ki so jih dobavile ali so jim bili dobavljeni, če ti pripomočki spadajo med pripomočke za vsaditev razreda III (9. odstavek 27. člena MDR in 24. člen IVDR). V skladu z 12. členom Zakona o medicinskih pripomočkih (ZMedPri-1) zdravstvene ustanove na enak način vodijo evidenco tudi za pripomočke, ki jim je priglašeni organ izdal certifikat in jih imajo v lasti. Zdravstvene ustanove vzpostavijo evidenco pripomočkov do 19. 6. 2026. Poslovni subjekti, ki pripomočke uporabljajo pri opravljanju svoje dejavnosti, pripomočke vzdržujejo v skladu z navodili proizvajalca pripomočkov.
V skladu s 20. členom Zakona o medicinskih pripomočkih (ZMedPri-1) imajo zdravstvene ustanove v svoji organizaciji vsaj eno osebo, ki je odgovorna za skladnost z zakonodajo, in vsaj eno osebo, ki je odgovorna za poročanje o vigilančnih zapletih. Naloge osebe, odgovorne za skladnost z zakonodajo, lahko opravlja tudi oseba, odgovorna za poročanje o vigilančnih zapletih. Oseba, odgovorna za skladnost z zakonodajo, poleg obveznosti iz drugega odstavka 14. člena Uredbe 2017/745/EU in drugega odstavka Uredbe 2017/746/EU vodi evidenco opravljenih nalog, ki predstavlja osnovo za izkazovanje izpolnjevanja njenih obveznosti. Osebe iz prvega stavka tega odstavka morajo imeti ustrezno strokovno znanje na področju medicinskih pripomočkov, ki se dokaže s potrdilom poslovnega subjekta na področju pripomočkov, ki izkazuje vsaj eno leto delovnih izkušenj z zakonodajnimi zahtevami ali sistemom vodenja kakovosti, povezanim z medicinskimi pripomočki, ali kopijo pogodbe oziroma drugim relevantnim dokumentom.
V skladu s 17. členom Zakona o medicinskih pripomočkih (ZMedPri-1) se morajo zdravstvene ustanove, ki izvajajo ponovno obdelavo pripomočkov za enkratno uporabo (v nadaljnjem besedilu: obdelovalci pripomočkov) v skladu s 14. členom tega zakona pred začetkom izvajanja te dejavnosti registrirati v informacijski sistem JAZMP.
V skladu z 18. členom Zakona o medicinskih pripomočkih (ZMedPri-1) se morajo zdravstvene ustanove, ki proizvajajo in uporabljajo pripomočke v skladu s 5. odstavkom 5. člena Uredbe 2017/745/EU, 5. odstavkom 5. člena Uredbe 2017/746/EU ter v skladu s 13. členom tega zakona pred začetkom izvajanja te dejavnosti registrirati v informacijski sistem JAZMP.
V skladu s 30. členom Zakona o medicinskih pripomočkih (ZMedPri-1) zdravstvena ustanova poslovnemu subjektu za namen preiskave v zvezi z resnim zapletom s pripomočkom omogoči dostop do pripomočka, pri katerem je prišlo do suma resnega zapleta in o katerem je poročala. Če to ni mogoče, zdravstvena ustanova zagotovi vzorec enakega pripomočka.
34. Kaj je EUDAMED, kako je sestavljen in kakšen je njegov namen?
Informacije so dostopne na povezavi:
https://www.jazmp.si/medicinski-pripomocki/splosno-o-medicinskih-pripomockih/eudamed/
35. Ali je registracija v EUDAMED obvezujoča?
V tem trenutku registracija v EUDAMED še ni obvezujoča. Registracija v EUDAMED bo obvezujoča 6 mesecev po objavi obvestila v Uradnem listu Unije, da eden ali več elektronskih sistemov (modulov) deluje in izpolnjuje funkcijske specifikacije.
Evropska Komisija in JAZMP že zdaj vzpodbujata vse gospodarske subjekte k registraciji v EUDAMED.
36. Kateri poslovni/gospodarski subjekti se registrirajo v EUDAMED in kateri v nacionalnih register pri JAZMP?
V EUDAMED se registrirajo proizvajalci, pooblaščeni predstavniki proizvajalca, uvozniki in proizvajalci sistema in paketa (22. člen MDR).
V nacionalni register pri JAZMP se do začetka delovanja Eudamed registrirajo vsi poslovni subjekti s sedežem v Republiki Sloveniji: proizvajalci, pooblaščeni predstavniki proizvajalca, distributerji, uvozniki in proizvajalci pripomočkov, izdelanih za posameznega uporabnika (custom made).
Po začetku delovanja Eudamed se bodo v nacionalni register pri JAZMP registrirali distributerji, proizvajalci medicinskih pripomočkov za posameznega uporabnika (custom made), obdelovalci pripomočkov in zdravstvene ustanove, ki proizvajajo in uporabljajo pripomočke v zdravstvenih ustanovah.
37. Kako poteka registracija poslovnega subjekta v EUDAMED in postopek pridobitve Enotne registrske številke poslovnega subjekta?
Poslovni subjekti v EUDAMED vnesejo informacije iz oddelka 1 dela A Priloge VI MDR in IVDR. Pristojni organ za medicinske pripomočke (v RS JAZMP) po preveritvi podatkov poslovnemu subjektu dodeli enotno registrsko številko (SRN – single registration number), ki izgleda kot je prikazano na sliki:

Proizvajalec enotno registrsko številko uporabi pri vložitvi vloge za ugotavljanje skladnosti pri priglašenem organu in za dostop do EUDAMED-a zaradi izpolnitve svojih obveznosti na podlagi 29. člena MDR in 26. člena IVDR.
38. Ali se morajo v nacionalni register pri JAZMP registrirati tudi lekarne?
V nacionalni register se morajo registrirati tudi lekarne in sicer kot distributerji (če pripomočkov ne uvažajo). Lekarne namreč z novo uredbo prevzemajo obveznosti distributerjev in morajo spoštovati vse določbe, ki veljajo za distributerje.
39. Kako je s prodajo na daljavo?
Prodaja na daljavo je definirana v 6. členu MDR, ki navaja, da mora biti pripomoček, ki je fizični ali pravni osebi s sedežem v Uniji na voljo prek storitev informacijske družbe, kot so opredeljene v točki (b) 1(1). člena Direktive 2015/1535 skladen z uredbo MDR oziroma IVDR.
40. Kako poteka registracija poslovnega/gospodarskega subjekta s sedežem v Republiki Sloveniji?
Registracija poslovnega/gospodarskega subjekta poteka preko portala Slovenske poslovne točke SPOT. Vlogo za registracijo lahko odda katera koli fizična oseba, ki je pooblaščena za oddajo vloge s strani zadevnega poslovnega subjekta in ima kvalificirano digitalno potrdilo.
Navodila za izpolnjevanje vloge so dostopne na povezavi:
https://spot.gov.si/assets/Navodila/Navodila-JAZMP.pdf
41. Kako je z registracijo poslovnega/gospodarskega subjekta s sedežem v Republiki Sloveniji v primeru, da podjetje NE razpolaga z kvalificiranim digitalnim potrdilom?
Navodila so dostopna na povezavi:
https://www.jazmp.si/medicinski-pripomocki/registracija-poslovnih-subjektov/
42. Kako se vrši registracija medicinskega pripomočka v RS?
Navodila so dostopna na povezavi:
https://www.jazmp.si/medicinski-pripomocki/registracija-medicinskih-pripomockov/
43. Ali so optiki proizvajalci medicinskih pripomočkov?
Velika večina optikov/očesnih ambulant v skladu z definicijo pripomočka, izdelanega za posameznega uporabnika in MDCG smernic MDCG 2021-3 Questions and Answers on Custom-Made Devices, tovrstnih pripomočkov ne šteje več med pripomočke, izdelane za posameznega uporabnika, ampak poslovne/gospodarske subjekte opredeljuje kot distributerje oz. uvoznike. Le-ti morajo izpolnjevati določbe 13. člena oziroma 14. člena MDR.
Smernica namreč pojasnjuje razlike med pripomočki, izdelanimi za posameznega uporabnika (angl. custom-made devices), medicinskimi pripomočki, prilegajočimi se uporabniku (angl. patient-matched devices) in pripomočki, prilagojenimi uporabniku (angl. adaptable devices).
Korekcijska očala, sestavljena s prilagoditvijo posameznih pripomočkov (okvir, stekla) največkrat niso pripomočki, izdelani za posameznega uporabnika v skladu z definicijo iz MDR, temveč so zgolj pripomočki, ki so bili prilagojeni v skladu z navodili proizvajalca.
44. Kako poteka postopek pridobitve Certifikata o prosti prodaji?
Navodila so dostopna na povezavi:
https://www.jazmp.si/medicinski-pripomocki/certifikat-o-prosti-prodaji
45. V kakšnem jeziku morajo biti napisana navodila za uporabo medicinskega pripomočka?
V skladu s 7. členom Zakona o medicinskih pripomočkih (ZMedPri-1) morajo biti v slovenskem jeziku oznake in navodila za uporabo iz oddelka 23 Priloge I Uredbe 2017/745/EU in oddelka 20 Priloge I Uredbe 2017/746/EU, ki so namenjene nestrokovnjaku, vključno z informacijami iz prvega odstavka 18. člena Uredbe 2017/745/EU (kartica o vsadku in formacije za paciente, ki imajo vsajen pripomoček).
Oznake in navodila za uporabo za pripomočke, ki jih proizvajalec opredeli za profesionalno uporabo, morajo biti v slovenskem ali drugem uporabniku razumljivem jeziku. Če navodilo za uporabo ni v slovenskem jeziku, mora poslovni subjekt, ki zdravstveni ustanovi dobavi pripomoček, na zahtevo zdravstvene ustanove takoj, najpozneje pa v desetih dneh od zahteve, posredovati navodilo za uporabo v slovenskem jeziku.
46. Ali mora biti navodilo za uporabo vključeno v prodajni embalaži medicinskega pripomočka?
Navodilo za uporabo mora biti vključeno v prodajni embalaži medicinskega pripomočka, razen za medicinske pripomočke razreda I in IIa, če jih je mogoče uporabljati varno tudi brez navodila za uporabo, vendar je treba podatke o medicinskem pripomočku zagotoviti uporabniku na njegovo zahtevo.
V ustrezno utemeljenih in izjemnih primerih navodila za uporabo za in vitro diagnostične medicinske pripomočke niso potrebna ali so lahko skrajšana, če se pripomoček brez takih navodil za uporabo lahko uporablja varno in kot je predvidel proizvajalec. Če je in vitro diagnostični medicinski pripomoček namenjen samo za strokovno uporabo, se navodila za uporabo lahko zagotovijo uporabniku v nepapirni obliki (npr. elektronski obliki), razen če je pripomoček predviden za testiranje ob pacientu. Če navodila za uporabo niso zagotovljena v papirnati obliki, je na oznaki pripomočka navedba o dostopnosti (ali razpoložljivosti) teh navodil, kadar je to ustrezno, pa tudi naslov spletnega mesta, kjer so na voljo.
47. Kaj mora vsebovati kartica o vsadku?
V skladu z 18. členom MDR mora kartica o vsadku, ki jo predloži proizvajalec, vsebovati informacije, ki omogočajo identifikacijo pripomočka, vključno z imenom pripomočka, serijsko številko, številko serije, UDI, modelom pripomočka ter imenom in naslovom proizvajalca ter njegovim spletnim mestom.
Vsebovati mora vsa opozorila, previdnostne ukrepe ali ukrepe, ki jih mora upoštevati pacient ali zdravstveni delavec glede vzajemne interference ob razumno predvidljivih zunanjih vplivih, zdravniških pregledih ali okoljskih razmerah.
Prav tako mora vsebovati vse informacije o pričakovani življenjski dobi pripomočka in vseh potrebnih nadaljnjih ukrepih, ter druge informacije, ki pacientu zagotavljajo varno uporabo pripomočka, vključno z informacijami iz točke (u) oddelka 23.4. Priloge I.
Več informacij in predpisana oblika kartice o vsadku je opredeljena v dveh smernicah:
MDCG 2021-11 – ”Guidance on Implant Card in – Device types”
MDCG 2019-8 v2 – Implant Card relating to the application of Article 18 Regulation (EU) 2017/745
Izgled kartice je predstavljen na spodnjih slikah. Prvi del podatkov na kartici o vsadku je pred-izpolnjen s strani proizvajalca, drugi del s podatki o bolniku izpolni zdravstvena ustanova.
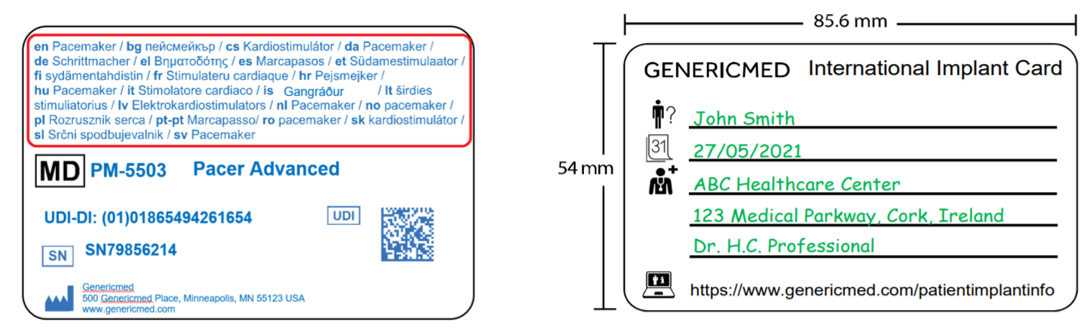
48. Kakšen je postopek dajanja na trg pripomočka v izrednih razmerah in kakšne so obveznosti gospodarskih subjektov?
Z odstopanjem od 52. člena MDR in od 48. člena IVDR lahko vsak pristojni organ na podlagi utemeljene zahteve na ozemlju zadevne države članice odobri dajanje določenega pripomočka, za katerega sicer niso bili opravljeni postopki iz navedenega člena, a je njegova uporaba v interesu varovanja javnega zdravja ali varnosti ali zdravja pacientov, na trg ali v uporabo.
Celoten postopek oddaje vloge in zahtevanih dokumentov v obravnavi vloge za izredno odobritev je dostopen na povezavi:
https://www.jazmp.si/medicinski-pripomocki/ostale-regulatorne-zadeve/neskladni-medicinski-pripomocki/
49. Kako ravnati z odpadki na področju medicinskih pripomočkov?
JAZMP nima pristojnosti nad ravnanjem z odpadki medicinskih pripomočkov.
V skladu z Uredbo 2017/745/EU in Uredbo 2017/746/EU je proizvajalec fizična ali pravna osebo, ki izdeluje ali popolnoma predela pripomoček ali naroči zasnovo, izdelavo ali popolno predelavo pripomočka in ki ta pripomoček trži pod svojim imenom ali blagovno znamko tisti, ki v svojih navodilih za uporabo kot tudi tehnični dokumentaciji opredeli ravnanje z nastalimi odpadki.
Prav tako ima svoje obveznosti distributer, ki omogoča dostopnost pripomočka. Te obveznosti so navedene v 14. členu obeh uredb.
Za lažje razumevanje smo na spletni strani JAZMP pripravili Priročnik za distributerje, ki je dostopen na spletni strani JAZMP. Priročnik povzema zahteve Uredbe 2017/745/EU (MDR) in Uredbe 2017/745/EU (IVDR). Za izpolnitev zahtev nacionalne zakonodaje upoštevajte določila Zakona o medicinskih pripomočkih (ZMedPri-1, Uradni list RS, št. 40/25).
50. Kakšna je definicija klinične raziskave v skladu z MDR?
Klinična raziskava medicinskega pripomočka je vsaka sistematična raziskava, ki vključuje eno ali več oseb, da se ocenita varnost ali učinkovitost pripomočka. Na tem mestu je potrebno opozoriti, da ta opredelitev vključuje tako uporabo pripomočkov, ki nimajo oznake CE, kot tiste s CE oznako.
Več informacij o kliničnih raziskavah je dostopnih na povezavi:
51. Kako poteka postopek priglasitve klinične raziskave?
Postopek priglasitve klinične raziskave je dostopen na povezavi:
https://www.jazmp.si/medicinski-pripomocki/klinicne-raziskave/postopek-priglasitve-pregleda-in-zakljucka-klinicne-raziskave/.
52. Kakšna je definicija študij učinkovitosti v skladu z IVDR in postopek priglasitve na JAZMP?
Študija učinkovitosti pomeni študijo, izvedeno za ugotavljanje ali potrjevanje analitične ali klinične učinkovitosti in vitro diagnostičnega medicinskega pripomočka.
Postopek priglasitve študije učinkovitosti je dostopen na povezavi:
https://www.jazmp.si/medicinski-pripomocki/studije-ovrednotenja-delovanja/postopek-priglasitve-pregleda-in-zakljucka-studije-ucinkovitosti/.
53. Kako je v skladu z uredbama opredeljena Vigilanca medicinskih pripomočkov?
Vigilančni sistem medicinskih pripomočkov je vzpostavljen z namenom zaščite javnega zdravja, zdravja in varnosti bolnikov ter ostalih uporabnikov medicinskih pripomočkov, z uvajanjem varnostno korektivnih ukrepov ter zmanjšanjem možnosti ponovitve resnega zapleta z medicinskim pripomočkom, ki se je v preteklosti že kdaj zgodil.
Aktivnosti, ki jih izvaja JAZMP v sistemu vigilance, so zbiranje in vrednotenje poročil o resnih zapletih v sodelovanju s proizvajalcem medicinskih pripomočkov ali pooblaščenim predstavnikom proizvajalca, spremljanje proizvajalca ali pooblaščenega predstavnika proizvajalca pri raziskovanju resnega zapleta, uvajanje katerih koli nadaljnjih ukrepov morda potrebnih za dopolnitev ukrepov proizvajalca ali pooblaščenega predstavnika ter sodelovanje v mednarodnem sistemu vigilance medicinskih pripomočkov.
54. Katere definicije na področju vigilance izhajajo iz MDR in IVDR?
„Zaplet“ pomeni vsako okvaro ali poslabšanje lastnosti ali učinkovitosti pripomočka, ki je dostopen na trgu, tudi zaradi napake ob uporabi, ki je posledica ergonomskih značilnosti, vsak neželen stranski učinek ter vsako neustrezno informacijo proizvajalca in vsako škodo, ki je posledica medicinske odločitve ali ukrepa, izvedenega ali neizvedenega na podlagi informacij ali rezultatov, pridobljenih s pripomočkom.
„Resni zaplet“ pa pomeni vsak zaplet, ki neposredno ali posredno povzroči ali bi lahko povzročil:
- smrt pacienta, uporabnika ali druge osebe,
- začasno ali trajno resno poslabšanje zdravstvenega stanja pacienta, uporabnika ali druge osebe,
- resno tveganje za javno zdravje.
„Resno tveganje za javno zdravje“ pomeni dogodek, katerega posledica je lahko neposredno tveganje smrti, resnega poslabšanja zdravstvenega stanja osebe ali hude bolezni, zaradi česar bi lahko bil potreben takojšen popravni ukrep, in ki lahko povzroči precejšnjo obolevnost ali umrljivost ljudi ali ki je neobičajen ali nepričakovan v določenem kraju in času.
Več informacij je dostopnih na povezavah:
https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/
55. Kdo poroča o zapletih z medicinskimi pripomočki?
O zapletih z medicinskimi pripomočki, ki se zgodijo na območju Republike Slovenije, JAZMP poročajo udeleženci v sistemu vigilance, kakor je opisano na povezavi: https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/porocanje-o-zapletih/.
Za lažje in hitrejše poročanje zdravstvenih delavcev, uporabnikov in pacientov o resnih zapletih smo na JAZMP pripravili tudi spletni obrazec, ki je dostopen na povezavi:
https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/porocanje-o-resnih-zapletnih-spletni-obrazec/
56. Na kakšen način in komu poročajo o resnem zapletu zdravstveni delavci, uporabniki in pacienti?
O zapletih z medicinskimi pripomočki, ki se zgodijo na območju Republike Slovenije, JAZMP poročajo udeleženci v sistemu vigilance, kakor je opisano na povezavi: https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/porocanje-o-zapletih/.
V kolikor ima zdravstveni delavec, uporabnik ali pacient kontakt proizvajalca ali distributerja, je pričakovano, da ga o nastalem zapletu obvesti, ključnega pomena pa je, da omogoči vračilo/pregled pripomočka, ki je povzročil zaplet (v primeru, da je na voljo tudi embalaža, se jo priloži pripomočku).
57. Na kakšen način in komu poročajo o resnem zapletu proizvajalci?
O zapletih z medicinskimi pripomočki, ki se zgodijo na območju Republike Slovenije, JAZMP poročajo udeleženci v sistemu vigilance, kakor je opisano na povezavi: https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/porocanje-o-zapletih/.
Dokler modul za vigilanco v EUDAMED-u še ni vzpostavljen, proizvajalec posreduje poročila o resnem zapletu, ki se je zgodil v Republiki Sloveniji, JAZMP z uporabo obrazcev, objavljenih na navedeni spletni strani JAZMP.
58. Na kakšen način in komu poročajo o resnem zapletu distributerji/uvozniki?
Distributerji in uvozniki, ki prejmejo pritožbe ali poročila o sumih na resne zaplete v zvezi s pripomočkom, katerega dostopnost so omogočili, te informacije takoj pošljejo proizvajalcu ter po potrebi pooblaščenemu predstavniku proizvajalca in uvozniku.
Če distributer ali uvoznik meni, da sum o resnem zapletu pomeni tudi resno tveganje, o tem obvesti tudi JAZMP, in sicer na obrazcu za poročanje o zaznanem resnem tveganju, kakor je opisano na https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/porocanje-o-zapletih/.
59. Poročanje JAZMP o resnem tveganju ali pojavu ponarejenih pripomočkov
V primeru zaznanega resnega tveganja v zvezi s pripomočkom ali suma, da je pripomoček ponarejen, uporabniki, pacienti, zdravstveni delavci ali gospodarski subjekti poročajo JAZMP na obrazcu o zaznanem resnem tveganju na elektronski naslov .
60. Na kakšen način in komu poročajo proizvajalci o resnem tveganju ali pojavu ponarejenih/ukradenih pripomočkov ?
Če proizvajalec meni, da pripomoček pomeni resno tveganje ali da je ponarejen/ukraden, o tem nemudoma obvesti pristojne organe držav članic, v katerih je omogočil dostopnost pripomočka.
JAZMP obvesti na obrazcu o zaznanem resnem tveganju (navede podrobnosti, zlasti o neskladnosti pripomočka in morebitnih sprejetih korektivnih ukrepih), ki ga pošlje na elektronski naslov .
61. Na kakšen način in komu poročajo distributerji/uvozniki o resnem tveganju ali pojavu ponarejenih/ukradenih pripomočkov (neskladnostih)?
Če distributer ali uvoznik meni, da pripomoček pomeni resno tveganje ali da je ponarejen/ukraden, o tem nemudoma obvesti proizvajalca in tudi pristojne organe držav članic, v katerih je omogočil dostopnost pripomočka.
JAZMP obvesti na obrazcu o zaznanem resnem tveganju (navede podrobnosti, zlasti o neskladnosti pripomočka in morebitnih sprejetih korektivnih ukrepih), ki ga pošlje na elektronski naslov .
62. Kako poteka poročanje pristojnim organom o varnostnih korektivnih ukrepih?
Proizvajalec medicinskih pripomočkov ali pooblaščeni predstavnik proizvajalca posreduje poročila o varnostnem korektivnem ukrepu vsem pristojnim organom držav članic Unije, v katerih je zadevni medicinski pripomoček dostopen na trgu in pristojnemu organu, v katerem ima proizvajalec ali pooblaščeni predstavnik proizvajalca sedež.
Navodila za poročanje so dostopna na povezavi:
https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/
63. Kdo je odgovoren za obveščanje uporabnikov z varnostnim obvestilom (obvestilo o varnostnem korektivnem ukrepu)?
Proizvajalec zagotovi, da so uporabniki zadevnega pripomočka nemudoma obveščeni o sprejetem varnostnem korektivnem ukrepu na podlagi uradnega, proizvajalčevega obvestila o varnostnem korektivnem ukrepu (varnostnega obvestila).
Več informacij je dostopnih na povezavi:
https://www.jazmp.si/medicinski-pripomocki/vigilanca-medicinskih-pripomockov/
64. Kakšna je vloga distributerjev/uvoznikov pri uvedbi varnostnega korektivnega ukrepa?
Distributerji sodelujejo s proizvajalcem ter po potrebi pooblaščenim predstavnikom proizvajalca in uvoznikom ter s pristojnimi organi, da bi zagotovili sprejetje potrebnih korektivnih ukrepov, na podlagi katerih bi pripomoček postal skladen ali bil umaknjen ali odpoklican s trga, kakor je ustrezno.
JAZMP pričakuje od distributerjev/uvoznikov čim hitrejše obveščanje/izvajanje ukrepa po njihovi distribucijski verigi, skladno z navodili proizvajalca znotraj varnostnega obvestila (obvestila o varnostnem korektivnem ukrepu). Datirana dokazila o izvedbi obveščanja/ izvajanja ukrepa morajo biti na voljo pristojnim organom, v kolikor jih zahtevajo, na vpogled.
65. Kateri priglašeni organi na območju EU so že imenovani za izdajanje certifikatov v skladu z MDR in IVDR in kje jih najdemo?
Že imenovani priglašeni organi za certificiranje pripomočkov v skladu z MDR in IVDR so dostopni na povezavi:
https://ec.europa.eu/growth/tools-databases/nando/.
66. Kako je v skladu z MDR definiran proizvajalec sistema ali paketa po 22. členu?
Fizične ali pravne osebe pripravijo izjavo, če pripomočke z oznako CE zaradi njihovega dajanja na trg v obliki sistema ali paketa kombinirajo z naslednjimi drugimi pripomočki ali izdelki, in sicer na način, ki je skladen s predvidenim namenom pripomočkov ali drugih izdelkov, in v mejah uporabe, ki jo določijo njihovi proizvajalci:
(a) drugimi pripomočki z oznako CE;
(b) in vitro diagnostičnimi medicinskimi pripomočki z oznako CE, ki so skladni z Uredbo 2017/746/EU;
(c) drugimi izdelki, ki so v skladu z zakonodajo, ki se uporablja za navedene izdelke, vendar samo, kadar se uporabljajo v medicinskem postopku ali kadar je njihova prisotnost v sistemu ali paketu drugače upravičena.
Zadevna fizična ali pravna oseba v izjavi iz odstavka 1 navede, da:
(a) je preverila medsebojno kompatibilnost pripomočkov in, če je to ustrezno, drugih izdelkov v skladu s proizvajalčevimi navodili ter izpeljala svoje dejavnosti v skladu z navedenimi navodili;
(b) je zapakirala sistem ali paket in priložila zadevne informacije za uporabnike, vključno z informacijami, ki jih zagotovijo proizvajalci pripomočkov in drugih izdelkov, ki so bili sestavljeni;
(c) je bila dejavnost kombiniranja pripomočkov in, če je to ustrezno, drugih izdelkov v en sistem ali paket izpeljana v skladu z ustreznimi metodami notranjega spremljanja, preverjanja in potrditve.
67. Kako je interni pripomoček opredeljen v IVDR in kakšne so zahteve za gospodarske subjekte?
Interni pripomoček opredeljen v IVDR je vsak IVD MP ali dodatek za IVD MP, ki se proizvaja in uporablja le v določeni zdravstveni ustanovi v skladu s 5. odstavkom 5. člena IVDR.
Z izjemo ustreznih splošnih zahtev glede varnosti in učinkovitosti, določenih v Prilogi I IVDR, se zahteve IVDR ne uporabljajo za pripomočke, ki se proizvajajo in uporabljajo le v zdravstvenih ustanovah s sedežem v Uniji, pod pogojem, da so izpolnjeni vsi naslednji pogoji:
(a) pripomočki niso preneseni na drug pravni subjekt,
(b) pripomočki so proizvedeni in se uporabljajo v okviru ustreznih sistemov vodenja kakovosti,
(c) laboratorij zdravstvene ustanove izpolnjuje zahteve iz standarda EN ISO 15189 ali po potrebi nacionalnih določb, tudi nacionalnih določb glede akreditacije,
(d) v dokumentaciji zdravstvene ustanove je utemeljeno, da specifičnih potreb ciljne skupine pacientov ni mogoče dovolj učinkovito zadovoljiti z enakovrednim pripomočkom, ki je dostopen na trgu,
(e) zdravstvena ustanova pristojnemu organu na zahtevo sporoči informacije o uporabi takšnih pripomočkov, kar vključuje tudi utemeljitev njihove proizvodnje, spremembe in uporabe,
(f) zdravstvena ustanova da izjavo, da bo med drugim objavila:
(i) naziv in naslov zdravstvene ustanove proizvajalke;
(ii) podatke, nujne za identifikacijo pripomočkov;
(iii) izjavo, da so pripomočki skladni s splošnimi zahtevami glede varnosti in učinkovitosti iz Priloge I k tej uredbi, in, kjer je to ustrezno, informacije o tem, katere zahteve niso v celoti izpolnjene, z ustrezno obrazložitvijo za to;
(g) glede pripomočkov razreda D v skladu s pravili iz Priloge VIII, zdravstvena ustanova pripravi dokumentacijo, iz katere je mogoče pridobiti informacije o proizvodnih prostorih, postopku izdelave, podatke o zasnovi in učinkovitosti pripomočkov, vključno s predvidenim namenom, in ki mora biti dovolj podrobna, da lahko pristojni organ oceni, ali so izpolnjene splošne zahteve glede varnosti in učinkovitosti iz Priloge I IVDR. Države članice lahko to določbo uporabijo tudi za pripomočke A, B ali C razreda v skladu s pravili iz Priloge VIII;
(h) zdravstvena ustanova z vsemi potrebnimi ukrepi zagotovi, da so vsi pripomočki proizvedeni v skladu z dokumentacijo iz točke (g), in
(i) zdravstvena ustanova preuči izkušnje, pridobljene pri klinični uporabi pripomočkov, ter sprejme vse potrebne korektivne ukrepe.
Nacionalne zahteve za zdravstvene ustanove, ki proizvajajo in uporabljajo pripomočke v zdravstvenih ustanovah, so opredeljene v 13. členu Zakona o medicinskih pripomočkih (ZMedPri-1).
Za interne pripomočke, ki jih zdravstvene ustanove proizvajajo za lastne potrebe in za katere so izpolnjeni pogoji iz petega odstavka 5. člena 2017/746/EU, zdravstvena ustanova objavi Izjavo o proizvodnji in uporabi internih pripomočkov iz točke (f) petega odstavka 5. člena Uredbe 2017/746/EU na svoji spletni strani, poleg tega pa jo lahko objavi tudi na javno dostopni oglasni deski v zdravstveni ustanovi.
Zdravstvena ustanova za interne pripomočke pripravi dokumentacijo iz točke (g) petega odstavka 5. člena Uredbe 2017/746/EU, in sicer poleg razreda D tudi za interne pripomočke razreda B in C. Organizacijska enota zdravstvene ustanove, ki proizvaja in vitro interne pripomočke, mora izpolnjevati najmanj zahteve iz standarda SIST EN ISO 15189.
JAZMP lahko, za namen preverjanja skladnosti, od zdravstvenih ustanov zahteva dokumentacijo iz prejšnjega odstavka in preostalo pripadajočo dokumentacijo. Od njih lahko zahteva celotno dokumentacijo o internih pripomočkih.
18. člen Zakona o medicinskih pripomočkih (ZMedPri-1) določa registracijo zdravstvenih ustanov, ki proizvajajo interne pripomočke.
68. Kaj so izdelki iz Priloge XVI?
To so izdelki brez predvidenega medicinskega namena, ki prav tako sodijo med pripomočke.
Izdelani morajo biti v skladu s skupnimi specifikacijami, ki so dostopne na povezavi: https://health.ec.europa.eu/medical-devices-sector/new-regulations_en.
Izdelki pred predvidenega medicinskega namena so v skladu z MDR razdeljeni v 6 skupin:
- Kontaktne leče ali drugi predmeti, namenjeni vnosu v ali na oko.
- Izdelki, namenjeni celotni ali delni vsaditvi v človeško telo s kirurško invazivnimi sredstvi za namen spremembe anatomije ali fiksacijo delov telesa, z izjemo izdelkov za tetoviranje in prebadanje telesa.
- Snovi, kombinacije snovi ali predmeti, ki se s podkožno, pod sluznično ali intradermalno injekcijo ali z drugim vnosom, uporabljajo kot obrazna ali ostala polnila za kožo ali sluznico, razen tisti za tetoviranje.
- Oprema, ki se uporablja za zmanjšanje, odstranjevanje ali uničevanje maščobnega tkiva, kot npr. oprema za liposukcijo, lipolizo ali lipoplastiko.
- Oprema, ki oddaja elektromagnetno sevanje visoke jakosti (infrardeča, vidna in ultravijolična svetloba) in je namenjena uporabi na človeškem telesu, vključno s koherentnimi in nekoherentnimi viri sevanja ter svetlobo monokromatskega in širokega spektra, npr. laserji in oprema z intenzivno pulzno svetlobo, ki se uporablja za obnavljanje krovnih plasti kože ali odstranjevanje vtetoviranih znamenj ali dlak ali drugo zdravljenje kože.
- Oprema za transkranialno stimulacijo možganov, ki uporablja električne tokove ali magnetna ali elektromagnetna polja, ki spreminjajo nevronsko delovanje možganov.
69. Ali je uvrstitev pripomočka na listo ZZZS v pristojnosti JAZMP?
JAZMP nima pristojnosti uvrstitve pripomočka na listo ZZZS. Zakonska obveznost vseh gospodarskih subjektov s sedežem v Republiki Sloveniji, ki se na kakršen koli način ukvarjajo z medicinskimi pripomočki je vpis v ustrezen register na JAZMP.
69. Ali izdaja JAZMP odločbe v angleškem jeziku?
Ne. JAZMP izdaja odločbe v skladu z zakonodajo v RS in sicer v slovenskem jeziku. Za prevod v angleški jezik JAZMP ni pristojna.
70. Kakšne pogoje morajo izpolnjevati medicinski pripomočki, ki so predmet javnih naročil?
JAZMP nima pristojnosti, da bi določala pogoje javnih naročil. Pogoje določa naročnik oziroma subjekt, ki izvaja javno naročilo.